Дарбэстим®
, растворРегистрационный номер
Торговое наименование
Международное непатентованное наименование
Лекарственная форма
раствор для инъекций
Состав
Действующее вещество в одном предварительно заполненном шприце Дарбэпоэтин альфа (рекомбинантный): 10 мкг (25 мкг/мл), 15 мкг (40 мкг/мл), 20 мкг (40 мкг/мл), 30 мкг (100 мкг/мл), 40 мкг (100 мкг/мл), 50 мкг (100 мкг/мл), 60 мкг (200 мкг/мл), 80 мкг (200 мкг/мл), 100 мкг (200 мкг/мл), 150 мкг (500 мкг/мл), 300 мкг (500 мкг/мл), 500 мкг (500 мкг/мл).
Вспомогательные вещества в 1 мл раствора: натрия дигидрофосфата моногидрат — 2,118 мг, натрия гидрофосфат — 0,661 мг, натрия хлорид — 8,182 мг, полисорбат 80 — 0,05 мг, вода для инъекций — до 1,0 мл.
Описание
Прозрачная, бесцветная жидкость.
Фармакотерапевтическая группа
Код АТХ
Фармакологические свойства
Дарбэпоэтин альфа производится с использованием генной технологии в клетках яичников китайского хомяка (СНО-К1).
Фармакодинамика
Дарбэпоэтин альфа стимулирует эритропоэз по тому же механизму, что и эндогенный эритропоэтин. Дарбэпоэтин альфа содержит пять N-связанных углеводных цепей, в то время как эндогенный гормон и рекомбинантные человеческие эритропоэтины (рчЭпо) имеют всего три цепи. Дополнительные остатки сахаров, с молекулярной точки зрения, не отличаются от таковых, представленных в эндогенном гормоне. Вследствие повышенного содержания углеводов дарбэпоэтин альфа обладает более длительным периодом полувыведения в сравнении с рчЭпо, а, следовательно, и большей активностью in vivo. Несмотря на указанные изменения молекулярной структуры дарбэпоэтин альфа сохраняет очень узкую специфичность к эритропоэтиновому рецептору.
Эритропоэтин — фактор роста, который в основном стимулирует образование эритроцитов. Рецепторы к эритропоэтину могут экспрессироваться на поверхности различных опухолевых клеток.
Больные с хронической почечной недостаточностью
В 2 клинических исследованиях было выявлено, что у пациентов с ХПН риск летального исхода и серьёзных сердечно-сосудистых нежелательных явлений выше при применении стимуляторов эритропоэза до более высоких целевых уровней гемоглобина при сравнении с более низкими (135 г/л (8,4 ммоль/л) против 113 г/л (7,1 ммоль/л); 140 г/л (8,7 ммоль/л) против 100 г/л (6,2 ммоль/л).
В рандомизированном, двойном слепом коррекционном исследовании (n = 358) сравнения режимов дозирования один раз каждые две недели и один раз в месяц у пациентов с хронической почечной недостаточностью, не находящихся на диализе, применение дарбэпоэтина альфа для коррекции анемии один раз в месяц было не хуже, чем один раз каждые две недели. Среднее время (1-й Квартиль, 3-й Квартиль) достижения коррекции уровня гемоглобина (≥100 г/л и ≥10 г/л по сравнению с исходным уровнем) составило 5 недель для обоих режимов дозирования (3,7 недель для режима дозирования один раз каждые две недели и 3,9 недель для режима дозирования один раз в месяц). В период оценки (недели 29–33), среднее значение (95 % ДИ) эквивалентной дозы составило 0,20 (0,17; 0,24) мкг/кг при режиме дозирования один раз каждые две недели и 0,27 (0,23; 0,32) мкг/кг при режиме дозирования один раз в месяц.
В рандомизированном, двойном слепом, плацебо-контролируемом исследовании (TREAT) 4038 пациентов с хронической почечной недостаточностью, с диабетом 2 типа и уровнем гемоглобина ≤110 г/л, не находящихся на диализе, получали дарбэпоэтин альфа с целью достижения уровня гемоглобина 130 г/л или плацебо (с назначением дарбэпоэтина альфа при снижении уровня гемоглобина ниже 90 г/л). Исследование не достигло основной цели, заключающейся в снижении риска смертности по любым причинам или по сердечно-сосудистой заболеваемости (дарбэпоэтин альфа vs плацебо; соотношение рисков 1,05; 95 % ДИ (0,94; 1.17), равно как и цели заключающейся в снижении смертности по любым причинам и прогрессирования до терминальной стадии почечной недостаточности (ТСПН) (дарбэпоэтин альфа vs плацебо; соотношение рисков 1,06; 95 % ДИ (0,95; 1,19). Анализ индивидуальных компонентов композитных конечных точек показал следующее соотношение рисков (95 % ДИ): летальный исход 1,05 (0,92; 1,21), хроническая сердечная недостаточность (ХСН) 0,89 (0,74; 1,08), инфаркт миокарда (ИМ) 0,96 (0,75; 1,23), инсульт 1,92 (1,38; 2,68), госпитализация в связи с ишемией миокарда 0,84 (0,55; 1,27), ТСПН 1,02 (0,87; 1.18).
Онкологические больные, получающие химиотерапию
Выживаемость и прогрессирование опухоли были изучены в общей сложности у 2833 пациентов в рамках пяти крупных контролируемых исследований. Из них четыре были двойными слепыми и плацебо-контролируемыми, а одно — открытым. В двух исследованиях включались больные, которым уже было проведено химиотерапевтическое лечение. В двух исследованиях целевой уровень гемоглобина устанавливался равным и выше 130 г/л, а в трёх других — в интервале от 120 до 140 г/л. В открытом исследовании не получено различий в показателях общей выживаемости между группой, получавшей лечение рчЭПО, и контрольной. В четырёх плацебо-контролируемых исследованиях показатели риска были в пользу контроля и находились в пределах от 1,25 до 2,47. В этих четырёх исследованиях был выявлен необъяснимый статистически достоверный прирост смертности по сравнению с контролем у больных с типичными видами рака и анемией, лечение которой проводилось рчЭПО. Сравнение частоты тромбозов и других осложнений в группах, получавших лечение рчЭПО, и контрольной, не даёт удовлетворительного объяснения причин этого прироста.
Также был проведён систематический анализ 57 исследований, включавших суммарно более 9000 онкологических пациентов. При мегаанализе общей выживаемости показатель риска равнялся 1,08 в пользу контроля (ДИ 95 %: 0,99-1,18; 8167 пациентов в 42 исследованиях).
У пациентов, получавших лечение рчЭПО, отмечалось повышение относительного риска развития тромбоэмболических событий (ОР = 1,67; ДИ 95 %: 1,35-2,06; 6769 пациентов в 35 исследованиях). Таким образом, существует достаточный объём данных, свидетельствующих о возможности возникновения значительного вреда при лечении онкологических больных рчЭПО. Неясно, до какой степени это применимо к случаям назначения рекомбинантных человеческих эритропоэтинов для достижения целевого уровня гемоглобина менее 130 г/л у пациентов с онкологическими заболеваниями, которые получают химиотерапию, поскольку в проанализированных данных имелось незначительное число пациентов с такими характеристиками.
Также был проведён анализ данных более чем у 13900 пациентов со злокачественными заболеваниями (химиотерапия, лучевая терапия, химиотерапия и лучевая терапия или отсутствие терапии), включённых в 53 контролируемых клинических исследования нескольких эпоэтинов. Метаанализ данных по общей выживаемости выявил соотношение рисков 1,06 в пользу контрольной группы (95 % ДИ: 1,00; 1,12; 53 исследования и 13933 пациента), и для пациентов со злокачественными заболеваниями, получающими химиотерапию, соотношение рисков общей выживаемости составило 1,04 (95 % ДИ: 0,97; 1,11; 38 исследований и 10441 пациента). Метаанализ также указывает на значительное повышение относительного риска тромбоэмболических событий у пациентов со злокачественными образованиями, получающих рекомбинантный человеческий эритропоэтин (см. раздел «Особые указания»).
Доклинические данные по безопасности
Во всех исследованиях на крысах и собаках при применении дарбэпоэтина альфа значимо возрастала концентрация гемоглобина, гемагокрита, эритроцитов и ретикулоцитов, что соответствует ожидаемому фармакологическому эффекту. Нежелательные явления при введении очень высоких доз препарата рассматривались как следствие усиленного фармакологического действия (снижения тканевого кровотока вследствие увеличения вязкости крови). Сюда же были отнесены миелофиброзы и гипертрофия селезёнки, а также расширение комплекса QRS на ЭКГ у собак, без нарушения сердечного ритма и влияния на интервал QT.
Дарбэпоэтин альфа не обладал каким-либо генотоксическим потенциалом и не оказывал влияния на пролиферацию клеток негематологического ряда ни in vitro, ни in vivo. В исследованиях по хронической токсичности не наблюдалось туморогенного или неожиданного митогенного ответа ни в одном изученном типе тканей. В продолжительных исследованиях на животных оценка канцерогенного потенциала дарбэиоэтина альфа не выполнялась.
В испытаниях, проводившихся на крысах и кроликах, не наблюдалось клинически значимого влияния на беременность, эмбриональное/фетальное развитие, роды или постнатальное развитие. Уровень проникновения препарата через плаценту был минимальным. Изменений фертильности не отмечалось.
Фармакокинетика
В связи с повышенным содержанием углеводов концентрация циркулирующего в крови дарбэпоэтина альфа превышает минимальную концентрацию необходимую для стимуляции эритропоэза в течение более продолжительного времени в сравнении с эквивалентными дозами рчЭпо, что позволяет снизить частоту введения дарбэпоэтина альфа с сохранением эквивалентного уровня биологического ответа.
Больные с хронической почечной недостаточностью
Фармакокинетика дарбэпоэтина альфа была изучена у больных с хронической почечной недостаточностью при внутривенном и подкожном введении препарата. Его период полувыведения составлял 21 час (стандартное отклонение (СО) 7,5) при внутривенном введении. Клиренс дарбэпоэтина альфа составил 1,9 мл/час/кг (СО 0,56), а объём распределения (Орс) был приблизительно эквивалентен объёму плазмы (50 мл/кг). При подкожном введении препарата его биодоступность соответствовала 37 %. При ежемесячном подкожном введении дарбэпоэтина альфа в дозе от 0,6 до 2,1 мкг/кг его период полувыведения составлял 73 часа (СО 24). Более продолжительный период полувыведения дарбэпоэтина альфа при подкожном введении, по сравнению с внутривенным, обусловлен кинетикой абсорбции. В ходе клинических исследований минимальное накопление препарата наблюдалось при любом способе введения. В доклинических исследованиях было продемонстрировано, что почечный клиренс дарбэпоэтина минимален (до 2 % общего клиренса) и не оказывает влияния на период полувыведения препарата из сыворотки.
Фармакокинетика дарбэпоэтина альфа изучалась у детей (3–16 лет) с ХПН, находящихся или не находящихся на диализе, при этом забор образцов проводился от момента однократного подкожного или внутривенного введения препарата до одной недели (168 часов) после введения. Периоды забора образцов были такой же продолжительности, как и у взрослых с ХПН, и сравнение показало, что фармакокинетика дарбэпоэтина альфа у взрослых и детей с ХПН похожа.
После внутривенного введения препарата отмечалось приблизительно 25 % различие между взрослыми и детьми в отношении площади под фармакокинетической кривой «концентрация–время» от нулевой отметки времени до бесконечности (AUC[0–∞]); тем не менее, указанное различие для детей составило менее двухкратного диапазона AUC[0–∞]. После подкожного введения препарата величина AUC[0–∞] у взрослых и детей была аналогичной. Как после внутривенного, так и после подкожного введения препарата, период полувыведения препарата у детей и взрослых с ХПН был сходен.
Онкологические больные, получающие химиотерапию
После подкожного введения препарата в дозе 2,25 мкг/кг взрослым онкологическим больным средние максимальные концентрации дарбэпоэтина альфа, составляющие 10,6 нг/мл (СО 5,9), устанавливались в среднем в течение 91 часа (СО 19,7). Эти параметры соответствовали линейной фармакокинетике дозы в широком диапазоне значений (от 0,5 до 8 мкг/кг при еженедельном введении от 3 до 9 мкг/кг при введении раз в две недели). Фармакокинетические параметры не изменялись при многократном дозировании в течение 12 недель (еженедельное введение или введение раз в две недели). Отмечалось ожидаемое умеренное повышение (<2-кратное) сывороточной концентрации препарата при достижении равновесного состояния, но не было выявлено признаков его накопления при повторном назначении.
Исследования фармакокинетики были выполнены с привлечением пациентов с индуцированной во время химиотерапии анемией, которые в комбинации с химиотерапией подкожно получали инъекции дарбэпоэтина альфа в дозе 6,75 мкг/кг раз в три недели. В данном исследовании среднее значение (СО) периода полувыведения составляло 74 (СО 27) часа.
Показания
- Лечение симптоматической анемии у взрослых и детей, страдающих хронической почечной недостаточностью (ХПН).
- Терапия симптоматической анемии у взрослых онкологических больных с немиелоидными злокачественными новообразованиями, получающих химиотерапию.
Противопоказания
- Повышенная чувствительность к дарбэпоэтину альфа, рчЭпо или к любому компоненту препарата.
- Неконтролируемая артериальная гипертензия.
С осторожностью
Заболевания печени; серповидно-клеточная анемия; эпилепсия.
Применение при беременности и в период грудного вскармливания
Полноценные, контролируемые исследования дарбэпоэтина альфа у беременных женщин не проводились.
В экспериментах на животных не было продемонстрировано прямого повреждающего действия препарата на течение беременности, на эмбриональное/фетальное развитие, на роды или постнатальное развитие. Влияния на фертильность выявлено не было.
При назначении препарата беременным женщинам следует соблюдать осторожность.
Неизвестно, экскретируется ли дарбэпоэтин альфа в грудное молоко. Поэтому не исключён риск для ребёнка при грудном вскармливании. Принятие решения о прекращении грудного вскармливания или прекращении/отмене лечения препаратом Дарбэстим® следует принимать, учитывая пользу грудного вскармливания для ребёнка и пользу лечения для матери.
Способ применения и дозы
Лечение дарбэпоэтином альфа должно проводиться врачами, имеющими опыт назначения по вышеупомянутым показаниям.
Дарбэстим® поставляется готовым для применения в предварительно заполненных шприцах (ПЗШ). Инструкции по применению препарата, обращению с ним и порядку его уничтожения приводятся в разделе «Особые указания по использованию».
Терапия симптоматической анемии в сочетании с хронической почечной недостаточностью (ХПН) у взрослых и детей
Симптомы анемии и последствия могут варьировать в зависимости от возраста пациентов, их пола и тяжести заболевания; в каждом случае необходим анализ индивидуальных клинических данных пациента лечащим врачом.
Дарбэпоэтин альфа может применяться подкожно или внутривенно для повышения уровня гемоглобина, но не выше 120 г/л. У больных, не находящихся на гемодиализе, подкожный способ введения является предпочтительным, так как позволяет избежать пункций периферических вен.
Уровень гемоглобина у пациентов подвержен индивидуальным колебаниям, в том числе иногда выше или ниже желаемых целевых значений. При отклонении уровня гемоглобина за пределы целевых значений проводят модификацию дозы, при этом под целевым значением следует рассматривать интервал от 100 до 120 г/л. Следует избегать стойкого повышения уровня гемоглобина выше 120 г/л, указания по коррекции дозы при повышении уровня гемоглобина выше 120 г/л представлены ниже. Также следует избегать повышения уровня гемоглобина более чем на 20 г/л за 4-недельный период. В этом случае также необходима коррекция дозы.
Лечение дарбэпоэтином альфа включает две стадии — фаза коррекции и поддерживающая фаза. Рекомендации по применению и дозированию у взрослых и детей в инструкции приводятся отдельно. Применение у детей в возрасте меньше 1 года не изучалось.
Взрослые пациенты с хронической почечной недостаточностью
Фаза коррекции
Начальная доза при подкожном или внутривенном введении должна составлять 0,45 мкг/кг массы тела при однократном еженедельном введении. Альтернативно, для больных, не получающих диализ, следующие начальные дозы препарата также могут назначаться подкожно: 0,75 мкг/кг массы тела каждые две недели или 1,5 мкг/кг массы тела один раз в месяц. Если повышение концентрации гемоглобина оказывается недостаточным (менее 10 г/л за 4 недели), доза препарата увеличивается приблизительно на 25 %. Повышение дозы препарата не должно осуществляться чаще, чем один раз в четыре недели.
Если увеличение содержания гемоглобина превышает 20 г/л за 4 недели, дозу препарата следует уменьшить примерно на 25 %. В случае, когда уровень гемоглобина превышает 120 г/л, следует рассмотреть возможность уменьшения дозы препарата. Если содержание гемоглобина продолжает увеличиваться, дозу следует снизить примерно на 25 %. Если после снижения дозы, гемоглобин продолжает повышаться, необходимо временно прекратить применение препарата до начала снижения уровня гемоглобина, после чего можно возобновить терапию, причём дозу препарата следует уменьшить примерно на 25 % от предыдущей дозы.
Гемоглобин следует измерять еженедельно или раз в две недели до его стабилизации. В последующем промежутки между измерениями гемоглобина можно увеличить.
Поддерживающая фаза
У пациентов, находящихся на диализе, можно продолжить вводить препарат Дарбэстим® один раз в неделю или перейти на введение один раз каждые две недели. При переводе пациентов, находящихся на диализе, с еженедельных инъекций на режим введения однократно раз в две недели, исходная доза должна вдвое превышать дозу, вводившуюся один раз в неделю. Для пациентов, не получающих диализа, можно продолжить вводить препарат Дарбэстим® один раз в неделю или один раз каждые две недели или один раз в месяц. Для пациентов, получающих препарат Дарбэстим® один раз каждые две недели, после достижения требуемой концентрации гемоглобина, его подкожное введение может производиться один раз в месяц с использованием исходной дозы вдвое превышающей предыдущую дозу, вводившуюся раз в две недели.
Титрование дозы с целью поддержания требуемой концентрации гемоглобина следует производить так часто, как это требуется.
Если для поддержания требуемого гемоглобина необходима оптимизация дозы препарата Дарбэстим®, её рекомендуется увеличивать приблизительно на 25 %. В случае, если наблюдается повышение гемоглобина более, чем 20 г/л за 4 недели, дозу препарата следует уменьшить приблизительно на 25 %, в зависимости от скорости повышения. Если содержание гемоглобина превышает 120 г/л, следует рассмотреть возможность уменьшения дозировки препарата. Если содержание гемоглобина продолжает увеличиваться, дозу следует снизить примерно на 25 %. Если после снижения дозы, гемоглобин продолжает повышаться, необходимо временно прекратить применение препарата до начала снижения уровня гемоглобина, после чего можно возобновить терапию, причём дозировку препарата следует уменьшить примерно на 25 % от предыдущей дозы.
Следует проводить тщательное наблюдение за пациентами для обеспечения адекватной коррекции анемии с применением минимальных одобренных доз препарата Дарбэстим®. После любого изменения дозы или режима введения, содержание гемоглобина следует контролировать каждую 1 или 2 недели. Изменение дозы во время поддерживающей фазы должно выполняться не чаще одного раза в две недели.
При изменении пути введения препарата следует использовать те же дозы препарата и осуществлять мониторинг концентрации гемоглобина раз в 1–2 недели с целью поддержания требуемого уровня гемоглобина.
Взрослых пациентов, получающих еженедельно по одной, две или три инъекции рчЭпо, можно перевести на режим однократного еженедельного введения препарата Дарбэстим® или его введение один раз в две недели. Исходную еженедельную дозу препарата Дарбэстим® (мкг/неделю) определяют, разделив общую еженедельную дозу рчЭпо (МЕ/неделю) на 200. Исходную дозу препарата Дарбэстим® (мкг/ в две недели) при режиме введения один раз в две недели определяют путём деления суммарной кумулятивной дозы рчЭпо, введённого за двухнедельный период, на 200. Ввиду известной индивидуальной вариабельности, для отдельных больных может потребоваться титрование доз до получения оптимального терапевтического эффекта. При замещении рчЭпо на препарат Дарбэстим® измерение уровня гемоглобина следует выполнять не реже одного раза в неделю или в две недели, а способ введения препарата должен оставаться неизменным.
Дети с хронической почечной недостаточностью
Фаза коррекции
Для детей в возрасте 11 лет и старше начальная доза при подкожном или внутривенном введении препарата составляет 0,45 мкг/кг веса тела в виде однократной инъекции один раз в неделю. У пациентов, не получающих диализ, может применяться начальная доза, равная 0,75 мкг/кг, подкожно один раз в две недели. Если увеличение уровня гемоглобина недостаточно (менее 10 г/л за 4-недельный период), необходимо увеличить дозу препарата примерно на 25 %. Увеличение дозы должно проводиться не чаще одного раза в четыре недели.
Если увеличение содержания гемоглобина превышает 20 г/л за 4 недели, дозу препарата следует уменьшить примерно на 25 % в зависимости от степени увеличения уровня гемоглобина. В случае, когда уровень гемоглобина превышает 120 г/л, следует рассмотреть возможность уменьшения дозы препарата. Если содержание гемоглобина продолжает увеличиваться, дозу следует снизить примерно на 25 %. Если после снижения дозы, гемоглобин продолжает повышаться, необходимо временно прекратить применение препарата до начала снижения уровня гемоглобина, после чего можно возобновить терапию, причём дозу препарата следует уменьшить примерно на 25 % от предыдущей дозы. Гемоглобин следует измерять еженедельно или раз в две недели до его стабилизации. В последующем промежутки между измерениями гемоглобина можно увеличить.
Лечение анемии препаратом Дарбэстим® в фазе коррекции у детей в режиме дозирования один раз в месяц не изучено.
Рекомендаций касательно коррекции уровня гемоглобина у детей в возрасте от 1 года до 10 лет нет.
Поддерживающая фаза
У детей 11 лет и старше в поддерживающую фазу терапии введение препарата Дарбэстим® можно продолжать в режиме один раз в неделю или один раз в две недели. Пациенты, находящиеся на диализе, при переводе их с режима дозирования препарата Дарбэстим® один раз в неделю в режим один раз в две недели первоначально должны получать дозу, эквивалентную удвоенной при однократном в неделю режиме введения. Если пациент не находится на диализе, после того, как достигнут целевой уровень гемоглобина в режиме дозирования препарата 1 раз в две недели, препарат Дарбэстим® может назначаться подкожно 1 раз в месяц, при этом начальная дозировка должна составлять удвоенную дозу от той, что использовалась в режиме 1 раз в две недели.
Для детей в возрасте от 1 года до 18 лет клинические данные показали, что пациенты, получающие рчЭпо два или три раза в неделю, могут быть переведены на препарат Дарбэстим®, вводимый 1 раз в неделю, и пациенты, получающие рчЭпо один раз в неделю, могут быть переведены на режим введения один раз в две недели. Начальная дозировка препарата Дарбэстим® для детей (мкг/нед), вводимого еженедельно может быть определена путём деления суммарной недельной дозы рчЭпо (МЕ/нед) на 240. Начальная дозировка препарата Дарбэстим® при введении каждые 2 недели (мкг/каждые 2 недели) может быть определена путём деления суммарной дозы рчЭпо за двухнедельный период на 240. По причине индивидуальных различий для отдельных пациентов требуется подбор оптимальной терапевтической дозы. При замене рчЭпо на препарат Дарбэстим®, уровень гемоглобина должен контролироваться каждые 1–2 недели, и при этом должен использоваться один и тот же способ введения препарата.
Титрование дозы с целью поддержания требуемой концентрации гемоглобина следует производить так часто, как это требуется.
Если для поддержания требуемого гемоглобина необходима оптимизация дозы препарата Дарбэстим®, её рекомендуется увеличивать приблизительно на 25 %.
Если увеличение содержания гемоглобина превышает 20 г/л за 4 недели, дозу препарата следует уменьшить примерно на 25 % в зависимости от степени увеличения уровня гемоглобина. В случае, когда уровень гемоглобина превышает 120 г/л, следует рассмотреть возможность уменьшения дозы препарата. Если содержание гемоглобина продолжает увеличиваться, дозу следует снизить примерно на 25 %. Если после снижения дозы, гемоглобин продолжает повышаться, необходимо временно прекратить применение препарата до начала снижения уровня гемоглобина, после чего можно возобновить терапию, причём дозу препарата следует уменьшить примерно на 25 % от предыдущей дозы.
Состояние пациентов следует тщательно мониторировать, для уверенности, что используемые минимальные одобренные дозы препарата Дарбэстим® обеспечивают адекватный контроль симптомов анемии.
После любого изменения дозы или режима введения, содержание гемоглобина следует контролировать каждую 1 или 2 недели. Изменение дозы во время поддерживающей фазы должно выполняться не чаще одного раза в две недели.
При изменении пути введения препарата следует использовать те же дозы препарата и осуществлять мониторинг концентрации гемоглобина раз в 1–2 недели с целью поддержания требуемого уровня гемоглобина.
Лечение симптоматической анемии, индуцированной химиотерапией, у пациентов с онкологическими заболеваниями
У пациентов с анемией (например, при концентрации гемоглобина равной или ниже 100 г/л) препарат Дарбэстим® может применяться подкожно для повышения уровня гемоглобина, но не выше 120 г/л. Симптоматика и последствия анемии зависят от возраста пациентов, их пола и тяжести заболевания. В каждом случае необходим анализ индивидуальных клинических данных пациента лечащим врачом.
Поскольку содержание гемоглобина в крови — индивидуальный показатель, для которого характерно выраженное разнообразие, у некоторых пациентов его содержание может как превышать целевой уровень, гак и быть меньше его. В этом случае помогает коррекция дозировки препарата, с учётом того, что целевой уровень гемоглобина составляет от 100 г/л до 120 г/л. Следует избегать повышения концентрации гемоглобина более 120 г/л; ниже представлено руководство по коррекции дозы в случае, если содержание гемоглобина превышает 120 г/л.
Рекомендованная начальная доза препарата — 500 мкг (6,75 мкг/кг) 1 раз в 3 недели либо по 2,25 мкг/кг 1 раз в неделю. Если клинический ответ (утомляемость, содержание гемоглобина) через девять недель неадекватен, дальнейшая терапия может оказаться неэффективной. Применение препарата Дарбэстим® прекращают примерно через четыре недели после завершения химиотерапии.
После достижения целевого уровня гемоглобина дозировку препарата следует уменьшить на 25–50 %, для адекватного контроля симптоматики анемии с использованием минимальных одобренных доз препарата Дарбэстим®. Возможно титрование дозы между 500 мкг, 300 мкг и 150 мкг.
Следует производить тщательный мониторинг состояния пациентов. Если уровень гемоглобина у пациента превышает 120 г/л, дозу препарата следует уменьшить на 25–50 %. Если содержание гемоглобина превышает 130 г/л, следует временно прекратить применение препарата Дарбэстим®. После снижения уровня гемоглобина до 120 г/л или ниже, терапию можно возобновить, дозировка препарата при этом должна быть примерно на 25 % меньше предыдущей.
Если увеличение уровня гемоглобина превышает 20 г/л за 4 недели, следует уменьшить дозировку препарата на 25–50 %.
Техника процедуры инъекции препарата Дарбэстим® в предварительно заполненных ширинах или в предварительно заполненных шприцах с защитным устройством для иглы
Этот раздел описывает процедуру инъекции препарата Дарбэстим®, которую вы можете выполнить самостоятельно. Перед началом применения препарата в предварительно заполненных шприцах и предварительно заполненных шприцах с защитным устройством для иглы, пожалуйста, сначала ознакомьтесь с «Общими рекомендациями», приведёнными ниже (раздел 1), а затем, с рекомендациями, приведёнными в разделе 2.
Раздел 1. Общие рекомендации
Очень важно, чтобы Вы не делали инъекцию сами, пока ваш лечащий врач, медицинская сестра или провизор не научит Вас. Если у Вас будут вопросы, то проконсультируйтесь с Вашим врачом, медицинской сестрой.
Перед началом инъекции
Внимательно прочитайте все рекомендации перед введением препарата.
Как Вам или тому, кто делает вам эту инъекцию, использовать ПЗШ?
Ваш врач назначил Вам препарат Дарбэстим® для подкожных инъекций. Ваш лечащий врач, медицинская сестра или провизор расскажет Вам какое количество препарата и как часто необходимо вводить.
Оборудование
Для самостоятельных инъекций вам потребуется:
- Один шприц (ПЗШ)/ ПЗШ с защитным устройством для иглы с препаратом Дарбэстим®;
- Спиртовая салфетка.
Перед проведением инъекции
1. Достать шприц из холодильника. Для извлечения предварительно заполненного шприца из пачки, возьмитесь за центр прозрачного защитного устройства для иглы.
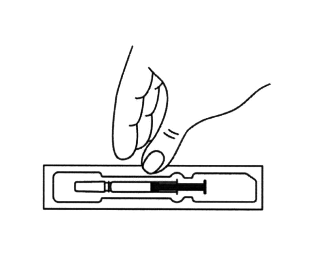
2. Оставить шприц при комнатной температуре примерно на 30 минут. Это позволит сделать инъекцию более комфортной.
3. Пожалуйста, обратите внимание на следующие инструкции:
a. Не подогревать предварительно заполненный шприц в горячей воде или в микроволновой печи.
b. Не оставлять заполненный шприц под солнечными лучами.
c. Не брать заполненный шприц за шток поршня, серый колпачок иглы или усики защитного устройства для иглы.
d. Не встряхивать шприц.
e. Не удалять колпачок предварительно заполненного шприца до тех пор, пока Вы не готовы к инъекции
f. Не пытайтесь активировать предварительно заполненный шприц перед инъекцией.
g. Не пытайтесь снять прозрачное защитное устройство для иглы с предварительно заполненного шприца.
Хранить предварительно заполненные шприцы в недоступном для детей месте.
4. Перед использованием необходимо проверить следующее:
a. Вы используете тот препарат и ту дозу, которые Вам назначил лечащий врач.
b. Срок годности на этикетке предварительно заполненного шприца (ГОДЕН ДО:). Не использовать шприц, если истёк последний день указанного месяца.
c. Описание препарата Дарбэстим®. Раствор должен быть прозрачным или бесцветным или. Если раствор мутный или содержит частицы, препарат использовать нельзя.
d. Вы можете заметить маленькие пузырьки воздуха в предварительно заполненном шприце. Вам не нужно удалять пузырьки воздуха перед инъекцией. Введение раствора с пузырьками воздуха является безопасным.
5. Тщательно вымыть руки.
6. Выбрать комфортное, хорошо освещённое место и чистую поверхность, удобно расположить все необходимые материалы.
Как выбрать место инъекции?
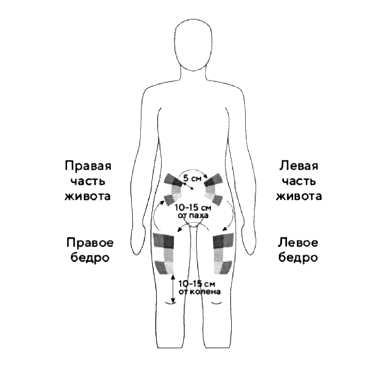
Лучше всего делать инъекции в верхнюю часть бёдер и живота. Если инъекции Вам делает кто-то другой, можно использовать тыльную поверхность рук.
Если область, куда Вы собрались делать инъекцию, покраснела или отекла, следует выбрать другое место инъекции.
Раздел 2. Рекомендации по введению препарата Дарбэстим
Как подготовиться к инъекции?
Перед инъекцией Вы должны сделать следующее:
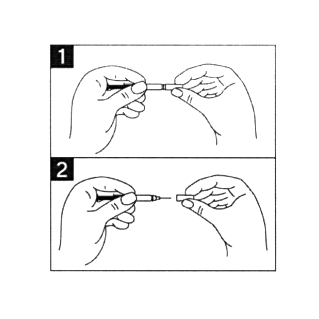
- Во избежание изгиба иглы, осторожно потянуть колпачок с иглы сразу, без скручивания, как показано на рисунках 1 и 2.
- Не дотрагиваться до иглы и не нажимать на поршень.
- Теперь Вы можете использовать предварительно заполненный шприц.
Как вводить препарат?
- Продезинфицировать место инъекции с помощью спиртовой салфетки, и зажать кожу (не сдавливая) большим и указательным пальцами.
- Ввести иглу в кожу полностью так, как показывал врач или медицинская сестра.
- Ввести назначенную дозу подкожно, как показывал Ваш врач или медицинская сестра.
- Медленно и непрерывно надавливать на поршень, при этом сжимать кожную складку и не отпускать её, пока шприц не опустеет.
- Извлечь иглу и отпустить складку кожи.
- Если выступит кровь, аккуратно вытереть её спиртовой салфеткой. Не растирать место инъекции. При необходимости, можно заклеить его пластырем.
- Один предварительно заполненный шприц предназначен для однократного применения. Не использовать оставшийся в шприце препарат Дарбэстим®.
Помните: При возникновении сложностей, обратитесь за помощью или советом к лечащему врачу или медицинской сестре.
В случае введения препарат Дарбэстим® в предварительно заполненном шприце с защитным устройством для иглы, Вам надо знать, что каждый шприц оснащён предохранителем иглы, который активируется автоматически для закрытия иглы после завершения инъекции.
Как вводить препарат Дарбэстим® в предварительно заполненном шприце с защитным устройством для иглы?
- Очистить кожу с помощью спиртовой салфетки.
- Во избежание загиба иглы, острожное потянуть колпачок с иглы сразу. Не дотрагиваться до иглы и не нажимать на поршень.
- Зажать кожу (не сдавливая) большим и указательным пальцами. Ввести иглу в кожу.
- Надавливать на поршень с лёгким непрерывным давлением. Надавливать на поршень следует до тех пор, пока шприц не опустеет. Прозрачный предохранитель иглы не сработает, пока предварительно заполненный шприц не опустеет. Пока поршень нажат, удалите иглу из кожи, отпустите поршень и позвольте шприцу подняться вверх до тех пор, пока вся игла не покроется прозрачным защитным устройством для иглы. Если защитное устройство для иглы не активизировалось, возможно, что Вы не выполнили инъекцию полностью. Позвоните Вашему лечащему врачу, если Вы считаете, что Вы не получили полную дозу.
- Если выступит кровь, аккуратно вытереть её спиртовой салфеткой. Не растирать место инъекции. При необходимости, можно заклеить его пластырем.
Побочное действие
Общие положения
Выявленные нежелательные реакции, связанные с приёмом дарбэпоэтина альфа, включают гипертензию, инсульт, тромбоэмболии, судороги, аллергические реакции, сыпь/эритему и парциальную красноклеточную аплазию (ПККА).
Боль в месте инъекции была зарегистрирована, как связанная с лечением, в исследованиях, в которых дарбэпоэтин альфа вводили в виде подкожных инъекций. Дискомфорт в месте инъекции в целом был слабым и преходящим и развивался преимущественно после первой инъекции.
Частота развития нежелательных реакций указана по классу системы органов и частоте возникновения.
Частота возникновения определена следующим образом: очень часто ≥1/10); часто (≥1/100, <1/10); нечасто (≥1/1 000, <1/100); редко (≥1/10 000, <1/1 000); очень редко (<1/10 000), неизвестно (не может быть оценена по имеющимся данным).
Данные представлены отдельно для пациентов с ХПН и онкологических пациентов и отражают профиль нежелательных реакций в этих популяциях.
Пациенты с хронической почечной недостаточностью
В контролируемых исследованиях из 1357 пациентов, 766 пациентов получали дарбэпоэтин альфа и 591 пациент получал рекомбинантный эритропоэтин человека. 83 % находились на диализе, 17 % — нет. Инсульт был выявлен в качестве нежелательной реакции в дополнительном клиническом исследовании (TREAT).
Частота нежелательных реакций, расценённых как связанные с лечением дарбэпоэтином альфа, составляет:
| Система органов по MedDRA | Частота возникновения | Нежелательная реакция |
|---|---|---|
| Со стороны крови и лимфатической системы | Неизвестна* | Парциальная красноклеточная аплазия |
| Со стороны иммунной системы | Очень часто* | Гиперчувствительность |
| Со стороны нервной системы | Часто | Инсульт |
| Нечасто* | Судороги | |
| Со стороны сердца | Очень часто | Повышение артериального давления |
| Со стороны сосудов | Редко | Тромбоэмболии |
| Со стороны кожи и подкожной клетчатки | Часто | Сыпь/Эритема |
| Со стороны организма в целом, включая местные реакции | Часто | Боль в месте инъекции |
* см. описание отмеченных реакций.
Онкологические больные
Нежелательные реакции были определены на основании объединённых данных семи рандомизированных двойных слепых плацебо-контролируемых исследований дарбэпоэтина альфа, включавших 2112 пациентов (дарбэпоэтин альфа 1200, плацебо 912). В клинические исследования включались пациенты с солидными опухолями (например, лёгких, молочной железы, толстой кишки, яичников) и лимфоидными злокачественными новообразованиями (например, лимфомой, множественной миеломой).
Частота нежелательных эффектов, расцененных как связанных с лечением дарбэпоэтином альфа, составляет:
| Система органов по MedDRA | Частота возникновения | Нежелательная реакция |
|---|---|---|
| Со стороны иммунной системы | Очень часто* | Гиперчувствительность |
| Со стороны нервной системы | Нечасто* | Судороги |
| Со стороны сердца | Часто* | Гипертензия |
| Со стороны сосудов | Часто | Тромбоэмболии, включая тромбоэмболию лёгочной артерии |
| Со стороны кожи и подкожной клетчатки | Часто | Сыпь/Эритема |
| Со стороны организма в целом, включая местные реакции | Очень часто | Отёк |
| Часто | Боль в месте инъекции |
*см. описание отмеченных нежелательных реакций.
Описание отмеченных нежелательных реакций
Пациенты с хронической почечной недостаточностью
Инсульт сообщался как распространённая нежелательная реакция у пациентов с ХПН в исследовании TREAT.
В отдельных случаях сообщалось о нейтрализующих антителах к эритропоэтину, опосредующих парциальную красноклеточную аплазию (ПККА), связанную с терапией дарбэпоэтином альфа, преимущественно у пациентов с ХПН, получавших препарат подкожно. В случае подтверждения диагноза ПККА, терапия дарбэпоэтином альфа должна быть прекращена, и пациенты не должны быть переведены на другой рекомбинантный эритропоэтин.
Частота реакций гиперчувствительности оценивалась на основе данных клинических исследований как «очень часто» у пациентов с ХПП. Сообщалось о развитии серьёзных реакций гиперчувствительности, включая анафилактические реакции, ангионевротический отёк, аллергический бронхоспазм, кожную сыпь и крапивницу, связанные с приёмом дарбэпоэтина альфа.
Сообщалось о судорогах у пациентов, получающих дарбэпоэтин альфа. Частота оценивалась на основе данных клинических исследований как «нечасто» у пациентов с ХПН.
Онкологические пациенты
При применении в рутинной клинической практике, у онкологических пациентов наблюдалась гипертензия. Частота оценивалась на основе данных клинических исследований как «часто» у онкологических пациентов и также часто встречалась в группах, получающих плацебо.
При применении в рутинной клинической практике, у онкологических пациентов наблюдались реакции гиперчувствительности. Частота реакций гиперчувствительности оценивалась на основе данных клинических исследований как «очень часто» у онкологических пациентов. Также реакции гиперчувствительности очень часто встречались в группах, получающих плацебо. Сообщалось о развитии серьёзных реакций гиперчувствительности, включающих анафилактические реакции, ангионевротический отёк, аллергический бронхоспазм, кожную сыпь и крапивницу, связанных с приёмом дарбэпоэтина альфа.
При применении в рутинной клинической практике у пациентов, получающих дарбэпоэтин альфа, сообщалось о судорогах. Частота оценивалась на основе данных клинических исследований как «нечасто» у онкологических пациентов. Судороги часто встречались в группах, получающих плацебо.
Дети с хронической почечной недостаточностью
Имеются ограниченные данные по безопасности применения дарбэпоэтина альфа у детей. Безопасность дарбэпоэтина альфа оценивалась в клиническом исследовании у детей с ХПН (от 1 года до 18 лет), получающих диализ, которые были стабильны при приёме эпоэтина альфа и затем, для поддержания уровня гемоглобина, были переведены на дарбэпоэтин альфа. Не было выявлено дополнительных нежелательных реакций у детей, по сравнению с ранее зарегистрированными у взрослых пациентов.
Передозировка
Максимальное количество дарбэпоэтина альфа, безопасное для введения в виде однократной или многократной дозы, не определялось. В случае недостаточного контроля уровня гемоглобина и титрования дозы, терапия дарбэпоэтином альфа может привести к полицитемии. После передозировки дарбэпоэтином альфа наблюдались случаи тяжёлой гипертензии.
В случае выявления полицитемии введение дарбэпоэтина альфа следует временно прекратить (см. раздел «Способ применения и дозы»). При наличии клинических показаний может быть выполнена флеботомия.
Взаимодействие с другими лекарственными средствами
Клинические данные, полученные до настоящего времени, не содержат указаний на взаимодействие дарбэпоэтина альфа с другими веществами. Однако известно, что потенциально возможно его взаимодействие с препаратами, характеризующимися высокой степенью сродства к эритроцитам, такими как циклоспорин, такролимус. При одновременном назначении дарбэпоэтина альфа с подобными лекарственными средствами следует контролировать уровень их содержания в сыворотке крови с модификацией дозы в случае повышения концентрации гемоглобина.
Ввиду того, что исследования по совместимости не проводились, дарбэпоэтин альфа не следует смешивать или вводить в виде инфузии вместе с другими медицинскими препаратами.
Особые указания
Общие положения
Необходим мониторинг артериального давления у всех пациентов, особенно в начале терапии препаратом Дарбэстим®. При не достижении адекватного контроля артериального давления стандартными методами, концентрация гемоглобина может быть снижена путём уменьшения дозы и отмены препарата Дарбэстим® (см. раздел «Способ применения и дозы»).
При лечении препаратом Дарбэстим® пациентов с ХПН, отмечалось развитие тяжёлой формы гипертензии, включая гипертонический криз, гипертоническую энцефалопатию и судорожные припадки.
С целью подтверждения эффективности эритропоэза, всем больным следует определять содержание железа до и во время лечения с целью назначения, в случае необходимости, дополнительной терапии препаратами железа.
Отсутствие ответа на применение препарата Дарбэстим® должно служить стимулом для выявления причинных факторов. Эффективность эритропоэз-стимулирующих препаратов (ЭСП) снижается при недостатке в организме железа, фолиевой кислоты или витамина B12, вследствие чего уровень их содержания необходимо корректировать. Эритропоэтический ответ также может быть ослаблен при наличии сопутствующих инфекционных заболеваний, симптомов воспаления или случаев травмы, скрытой кровопотери, гемолиза, тяжёлой алюминиевой интоксикации, сопутствующих гематологических заболеваний или фиброза костного мозга. Численность ретикулоцитов следует рассматривать как один из параметров оценки. Если типичные причины отсутствия ответа исключены, а у больного выявляется ретикулоцитопения, следует провести исследование костного мозга. Если картина костного мозга соответствует картине ПККА, рекомендуется выполнить исследование на присутствие антител к эритропоэтину.
Была описана ПККА, вызванная нейтрализующим действием антиэритропоэтиновых антител, связанная с применением ЭСП, включая и дарбэпоэтин альфа. Чаще всего такие сообщения касались пациентов с ХПН, получавших препарат подкожно. Было показано, что эти антитела перекрёстно реагируют со всеми эритропоэтическими белками. В случае постановки диагноза ПККА, лечение препаратом дарбэпоэтина альфа необходимо прекратить без последующего перевода на терапевтический режим, включающий другой рекомбинантный эритропоэтический белок (см. раздел «Побочное действие»). Парадоксальное снижение уровня гемоглобина и развитие тяжёлой анемии с низким уровнем ретикулоцитов должно приводить к немедленной отмене лечения эпоэтином и проведению теста на наличие антител к эритропоэтину. Такие случаи были описаны у пациентов с гепатитом C, которые получали терапию интерфероном и рибавирином в сочетании с эпоэтинами. Применение эпоэтинов в лечении анемии при гепатите C не одобрено.
Во всех исследованиях дарбэпоэтина альфа критерием исключения были активные заболевания печени, поэтому данные о применении препарата у больных с нарушением функции печени отсутствуют. Так как печень считается основным путём выведения рчЭпо, пациентам с патологией печени эти препараты следует назначать с осторожностью. Злоупотребление дарбэпоэтином альфа у здоровых лиц может привести к избыточному повышению гематокрита. Подобные явления могут быть ассоциированы с опасными для жизни осложнениями со стороны сердечно-сосудистой системы.
При поддержании уровня гемоглобина у пациентов с хронической почечной недостаточностью, его концентрация не должна превышать верхней границы, указанной в разделе «Способ применения и дозы». В ходе клинических исследований, при достижении целевого уровня гемоглобина более 120 г/л на фоне применения ЭСП, у пациентов отмечался повышенный риск смертности, развития серьёзных осложнений со стороны сердечно-сосудистой системы или нарушений мозгового кровообращения, включая инсульт и тромбоз сосудистого доступа.
В ходе контролируемых клинических исследований не удалось выявить значительных преимуществ от применения эпоэтинов, если концентрация гемоглобина превышает уровень, необходимый для контроля симптомов анемии и устранения потребности в гемотрансфузиях.
Дарбэстим® следует применять с осторожностью у пациентов с эпилепсией. Имеются сообщения о возникновении судорог у пациентов, получавших дарбэпоэтин альфа.
Больные с хронической почечной недостаточностью
У пациентов с хронической почечной недостаточностью, поддерживающие концентрации гемоглобина не должны превышать верхнюю границу целевой концентрации гемоглобина, рекомендованной в разделе «Способ применения и дозы». В ходе клинических исследований наблюдались повышенные риски летального исхода, серьёзных сердечно-сосудистых осложнений или нарушений мозгового кровообращения, включая инсульт, и тромбоз сосудистого доступа при назначении ЭСП для достижения уровня гемоглобина свыше 120 г/л (7,5 ммоль/л).
Проводимые контролируемые клинические исследования не показали значительных преимуществ при назначении эпоэтинов, когда концентрация гемоглобина повышается выше уровня, необходимого для лечения симптомов анемии и для того, чтобы избежать переливания крови.
Всем пациентам с уровнем ферритина сыворотки крови ниже 100 мкг/л или тем, у кого насыщение трансферрина ниже 20 %, рекомендуется дополнительное лечение препаратами железа.
Во время применения препарата Дарбэстим® следует регулярно контролировать сывороточное содержание калия. Повышение концентрации калия было описано у нескольких пациентов, получающих дарбэпоэтин альфа, однако причинная связь установлена не была. При выявлении повышенной или повышающейся концентрации калия, введение дарбэпоэтина альфа следует прекратить до её нормализации.
Онкологические больные
Влияние на рост опухоли
Эпоэтины представляют собой факторы роста, которые, главным образом, стимулируют выработку эритроцитов. Рецепторы к эритропоэтину могут экспрессироваться на поверхности различных опухолевых клеток. Как и в случае любых факторов роста, существует предположение о том, что эритропоэтины способны стимулировать рост опухолей.
В ряде контролируемых клинических исследований у онкологических больных, получающих химиотерапию, применение эпоэтинов не увеличивало общую продолжительности жизни или не снижало риск прогрессии опухоли у пациентов, с анемией, ассоциированной с онкологическим заболеванием.
В контролируемых клинических исследованиях дарбэпоэтина альфа и других ЭСП было продемонстрировано:
- Уменьшение времени до прогрессии у пациентов с распространённым раком головы и шеи, получающими лучевую терапию, при корректирующем назначении эпоэтина до достижения целевого значения гемоглобина выше, чем 140 г/л, ЭСП не показаны данной группе пациентов.
- Уменьшение общей продолжительности жизни и повышение смертности, связанное с прогрессией заболевания за 4 месяца у пациентов с метастазирующим раком молочной железы, получавших химиотерапию, при корректирующем назначении эпоэтина до достижения, целевого значения гемоглобина 120–140 г/л.
- Повышение риска смерти при корректирующем назначении эпоэтина до достижения целевого значения гемоглобина 120 г/л у пациентов с активной злокачественной опухолью, не получавших ни химиотерапии, ни лучевой терапии. Эритропоэз-стимулирующие агенты не показаны данной группе пациентов.
В соответствии с вышеизложенным, в некоторых клинических ситуациях для лечения анемии у пациентов с онкологическими заболеваниями следует применять переливание крови. Решение о назначении рекомбинантных эритропоэтинов следует принимать на основании оценки соотношения польза/риск для каждого индивидуального пациента, принимая во внимание особенности клинической ситуации. Необходимо учитывать следующие факторы: вид и стадия опухолевого процесса; степень анемии; ожидаемая продолжительность жизни; обстановка, в которой пациент будет проходить лечение; и пожелания самого пациента (см. раздел «Фармакодинамика»).
У больных с солидными опухолями или с лимфопролиферативными злокачественными заболеваниями при возрастании уровня содержания гемоглобина выше 120 г/л следует, строго соблюдать схему адаптации дозы, описанную в разделе «Способ применения и дозы», с целью минимизации потенциального риска развития тромбоэмболических явлений. Также необходимо регулярно контролировать численность тромбоцитов и концентрацию гемоглобина в крови.
Особые указания по использованию
Дарбэпоэтин альфа представляет собой стерильный продукт, изготовленный без консервантов.
Одним шприцом следует вводить не более одной дозы препарата. Любое количество лекарственного препарата, оставшееся в предварительно заполненном шприце, подлежит уничтожению.
Перед введением раствор препарата Дарбэстим® следует проконтролировать на предмет присутствия видимых частиц. Допускается использование только бесцветного, прозрачного или слабо опалесцирующего раствора. Раствор нельзя встряхивать. Перед введением следует дождаться прогревания ПЗШ до комнатной температуры.
Чтобы избежать возникновения дискомфорта в месте инъекции, необходимо менять места введения препарата.
Любые количества неиспользованного препарата или его отходов подлежат уничтожению в соответствии с местными требованиями.
Влияние на способность управлять транспортными средствами, механизмами
Влияние дарбэпоэтина альфа на способность к вождению автомобиля и обращению с техникой выявлено не было.
Форма выпуска
Раствор для инъекций, 25 мкг/мл, 40 мкг/мл, 100 мкг/мл, 200 мкг/мл и 500 мкг/мл.
Первый вариант упаковки:
Предварительно заполненные шприцы по 0,4 мл раствора (25 мкг/мл) — 10 мкг, или 0,375 мл раствора (40 мкг/мл) — 15 мкг, или 0,5 мл раствора (40 мкг/мл) — 20 мкг или 0,3 мл раствора (100 мкг/мл) — 30 мкг, или 0,4 мл раствора (100 мкг/мл) — 40 мкг, или 0,5 мл раствора (100 мкг/мл) — 50 мкг, или 0,3 мл раствора (200 мкг/мл)- 60 мкг, или 0,4 мл раствора (200 мкг/мл) — 80 мкг, или 0,5 мл раствора (200 мкг/мл) -100 мкг, или 0,3 мл раствора (500 мкг/мл) — 150 мкг или 0,6 мл раствора (500 мкг/мл) — 300 мкг, или 1,0 мл раствора (500 мкг/мл) — 500 мкг в шприцы из стекла 1 гидролитического класса.
На каждый шприц наклеивают этикетку.
По 1 шприцу помещают в контурную ячейковую упаковку из плёнки ПВХ. По 1 или 4 контурные ячейковые упаковки (по 0,3; 0,375; 0,4 или 0,5 мл препарата) или по 1 контурной ячейковой упаковке (по 0,6 или 1,0 мл препарата) помещают вместе с инструкцией по применению в пачку из картона. Пачка со шприцами дополнительно комплектуется спиртовыми салфетками в количестве 1 или 4 шт.
Второй вариант упаковки:
Предварительно заполненные шприцы по 0,4 мл раствора (25 мкг/мл) — 10 мкг, или 0,375 мл раствора (40 мкг/мл) — 15 мкг, или 0,5 мл раствора (40 мкг/мл) — 20 мкг или 0,3 мл раствора (100 мкг/мл) — 30 мкг, или 0,4 мл раствора (100 мкг/мл) — 40 мкг, или 0,5 мл раствора (100 мкг/мл) — 50 мкг, или 0,3 мл раствора (200 мкг/мл)- 60 мкг, или 0,4 мл раствора (200 мкг/мл) — 80 мкг, или 0,5 мл раствора (200 мкг/мл) -100 мкг, или 0,3 мл раствора (500 мкг/мл) — 150 мкг или 0,6 мл раствора (500 мкг/мл) — 300 мкг, или 1,0 мл раствора (500 мкг/мл) — 500 мкг в шприцы из стекла 1 гидролитического класса.
На каждый шприц наклеивают этикетку.
По 1 шприцу с системой защиты иглы помещают в контурную ячейковую упаковку из плёнки ПВХ. По 1 или 4 контурные ячейковые упаковки (по 0,3; 0,375; 0,4 или 0,5 мл препарата) или по 1 контурной ячейковой упаковке (по 0,6 или 1,0 мл препарата) помещают вместе с инструкцией по применению в пачку из картона. Пачка со шприцами дополнительно комплектуется спиртовыми салфетками в количестве 1 или 4 шт.
Хранение
Хранить при температуре 2–8 °C, в защищённом от света месте. Не замораживать.
Хранить в недоступном для детей месте.
Срок годности
3 года.
Не использовать по окончанию срока годности.
Условия отпуска из аптек
Отпускают по рецепту
Производитель
БИОКАД, ЗАО, Российская Федерация
Подробнее по теме
Ознакомьтесь с дополнительными сведениями о препарате Дарбэстим: