Кайендра
, таблеткиРегистрационный номер
Торговое наименование
Кайендра
Международное непатентованное наименование
Лекарственная форма
таблетки, покрытые плёночной оболочкой
Состав
Действующее вещество: сипонимода фумарат (сипонимод и фумаровая кислота, сокристалл) (Сипонимод) — 0,278 (0,250) мг или 2,224 (2,000) мг;
вспомогательные вещества: лактозы моногидрат, целлюлоза микрокристаллическая (pH 102), кросповидон (тип А), глицерил дибегенат, кремния диоксид коллоидный;
оболочка плёночная: оболочка Premix белая (поливиниловый спирт частично гидролизованный, титана диоксид (E171), тальк, лецитин соевый (E322), камедь ксантановая), оболочка Premix красная (поливиниловый спирт частично гидролизованный, краситель железа оксид красный (E172), тальк, лецитин соевый (E322), камедь ксантановая), для дозировки 0,25 мг: оболочка Premix чёрная (поливиниловый спирт частично гидролизованный, краситель железа оксид чёрный (E172), тальк, лецитин соевый (E322), камедь ксантановая), для дозировки 2 мг: оболочка Premix жёлтая (поливиниловый спирт частично гидролизованный, краситель железа оксид жёлтый (E172), тальк, лецитин соевый (E322), камедь ксантановая).
Описание
Дозировка 0,25 мг: круглые, двояковыпуклые таблетки, покрытые плёночной оболочкой светло-красного цвета, со скошенными краями, без риски, с гравировкой «Т» на одной стороне и «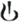 » — на другой.
» — на другой.
Дозировка 2 мг: круглые, двояковыпуклые таблетки, покрытые плёночной оболочкой светло-жёлтого цвета, со скошенными краями, без риски, с гравировкой «II» на одной стороне и «» — на другой.
Фармакотерапевтическая группа
Код АТХ
Фармакологические свойства
Фармакодинамика
Механизм действия
Сипонимод является модулятором рецептора сфингозин-1-фосфата (S1P). Он селективно связывается с двумя из пяти типов рецепторов S1P, сопряжённых с G-белком (G-protein-coupled receptors, GPCR), а именно S1P1 и S1P5. Выступая как функциональный антагонист рецепторов S1P1 на поверхности лимфоцитов, сипонимод препятствует их выходу из лимфатических узлов. Это приводит к снижению рециркуляции Т-лимфоцитов в центральную нервную систему (ЦНС) и, таким образом, ограничивает развитие центрального воспаления. Сипонимод не оказывает влияния на эффекторные Т-клетки памяти в периферических тканях и крови и не нарушает активацию лимфоцитов.
Сипонимод легко проникает через гематоэнцефалический барьер.
В исследованиях у животных показано прямое действие сипонимода на нервные клетки путём воздействия на S1P1 на астроцитах и S1Р5 на олигодендроцитах. На мышиной модели экспериментального аутоиммунного энцефаломиелита показан прямой нейропротекторный эффект сипонимода при его центральном введении (путём инфузий в желудочковую систему головного мозга), не зависящий от его влияния на лимфоциты.
Клиническая эффективность
Эффективность сипонимода была продемонстрирована в исследовании III фазы при его применении в дозе 2 мг в сутки у пациентов с вторично прогрессирующим рассеянным склерозом (ВПРС).
Исследование А2304 EXPAND представляло собой рандомизированное двойное слепое плацебо-контролируемое исследование III фазы продолжительностью, зависящей от количества исходов и длительности последующего наблюдения у пациентов с ВПРС с документальными признаками прогрессии заболевания за последние 2 года при отсутствии рецидивов или вне зависимости от таковых, без признаков рецидива на протяжении 3 месяцев перед включением в исследование, и средней оценкой от 3,0 до 6,5 баллов по шкале EDSS (Expanded Disability Status Scale — расширенная шкала оценки степени инвалидизации) на момент включения в исследование.
Первичной конечной точкой исследования был период времени до наступления 3-месячного подтверждённого прогрессирования нетрудоспособности (ППН), определяемого как увеличение значения EDSS минимум на 1 балл от исходного (увеличение на 0,5 балла для пациентов с исходным EDSS 5,5 или более), сохраняющееся в течение 3 месяцев.
Основными вторичными конечными точками были время до подтверждённого ухудшения оценки в тесте ходьбы на 25 футов как минимум на 20 % и изменение объёма поражения на T2-взвешенных изображениях.
В исследовании 1651 пациент был рандомизирован в группу сипонимода с приёмом в дозе 2 мг (N = 1105) или плацебо (N = 546); 82 % пациентов, получавших сипонимод, и 78 % пациентов, получавших плацебо, завершили исследование. Медиана возраста составила 49 лет, медиана продолжительности заболевания — 16 лет, а медиана исходной оценки по EDSS — 6,0. У 64 % пациентов в течение 2 лет до включения в исследование не было рецидивов. У 76 % по данным исходной магниторезонансной томографии (МРТ) не обнаружено очагов с контрастным усилением гадолинием. До включения в исследование 78 % пациентов ранее получали терапию по поводу PC.
На фоне приёма сипонимода статистически значимо увеличилось время до начала ППН (основная конечная точка), при этом по сравнению с плацебо риск снизился на 21,2 % (отношение рисков (ОР) 0,79, р <0.0134).
Результаты исследования указывали на варьирующее, но стойкое снижение риска 3- и 6-месячного ИПН при применении сипонимода по сравнению с плацебо в подгруппах пациентов, сформированных по полу, возрасту, наличию обострений до начала исследования, активности заболевания по результатам исходной МРТ, длительности заболевания и исходному уровню инвалидизации.
Применение сипонимода не привело к статистически значимому увеличению времени до подтверждённого ухудшения на ≥20 % в тесте ходьбы на 25 футов по сравнению с плацебо (численное снижение риска составило 6,2 %).
Периоды до наступления 3- и 6-месячного ППН были значительно продолжительнее у пациентов с активным заболеванием, получавших сипонимод, соответственно на 31 % (ОР 0,69; 95 % ДИ: 0,53, 0,91) и 37 % по сравнению с плацебо (ОР 0,63; 95 % ДИ: 0,47, 0,86). По сравнению с плацебо подтверждённая годовая частота обострений (ARR, annualized relapse rate) снизилась на 46 % (отношение ARR 0,54; 95 % ДИ: 0,39, 0,77). Относительное снижение частоты совокупного количества T1-взвешенных очагов с контрастным усилением гадолинием за 24 месяца составило 85 % (соотношение частоты 0,155; 95 % ДИ: 0,104, 0,231) по сравнению с плацебо. Различия в изменении объёма поражения на T2 и в процентном изменении объёма головного мозга (в среднем через 12 и 24 месяца) по сравнению с плацебо составляли -1163 мм3 (95 % ДИ: -1484, -843 мм3) и 0,141 % (95 % ДИ: 0,020, 0,261 %) соответственно.
Иммунная система
В течение 6 часов после приёма первой дозы сипонимод вызывает дозозависимое снижение количества лимфоцитов в периферической крови из-за обратимого перераспределения в лимфоидных тканях.
На фоне продолжительного ежедневного приёма количество лимфоцитов продолжает снижаться, достигая наименьшего срединного значения (90 % ДИ), приблизительно равного 0,560 (0,271–1,08) клеток/нл, что составляет 20–30 % от исходного уровня у типичных пациентов с ВПРС не японского происхождения с генотипом изофермента СYP2C9*1*1 или *1*2. Низкое количество лимфоцитов сохраняется при продолжительном ежедневном приёме.
У подавляющего большинства (90 %) пациентов с ВПРС количество лимфоцитов, как правило, возвращается к норме в течение 10 дней после прекращения приёма препарата. Остаточный эффект снижения количества лимфоцитов в периферической крови может сохраняться в течение 3–4 недель после приёма последней дозы препарата.
Электрофизиология сердца
Ритм и частота сердечных сокращений
В начале приёма сипонимод вызывает временное снижение частоты сердечных сокращений (ЧСС) и замедление атриовентрикулярной проводимости (см. раздел «Побочное действие»). Максимальное снижение ЧСС наблюдается в первые 6 часов после приёма препарата. Приём сипонимода не оказывает отрицательного влияния на вегетативные реакции сердца, включая циркадную вариабельность ЧСС и ответ на физическую нагрузку. В группе сипонимода в начальной фазе терапии по сравнению с плацебо чаще наблюдались временное дозозависимое снижение ЧСС, которое достигало плато при применении доз ≥5 мг, а также более высокая частота возникновения брадиаритмических нарушений (АВ-блокада и синусовая пауза).
Не зарегистрировано случаев атриовентрикулярной блокады (АВ-блокады) второй степени типа Мобитц II или более высокой степени. В большинстве случаев АВ-блокада и синусовая пауза возникали при применении дозы, превышающей терапевтическую 2 мг, с заметно более высокой частотой возникновения таких случаев при отсутствии фазы титрации (пошаговое увеличение дозы). Снижение ЧСС, вызванное сипонимодом, может быть скорректировано применением атропина или изопреналина.
Потенциал к удлинению интервала QT
Влияние сипонимода на реполяризацию миокарда в терапевтических (2 мг) и супратерапевтических дозах (10 мг) было оценено в специализированном QT-исследовании. Не получено данных о наличии у сипонимода аритмогенного потенциала, связанного с удлинением интервала QT. Через 3 часа после приёма дозы сипонимода увеличивалось среднее плацебо-скорректированное значение QTcF (ΔΔQTcF) с поправкой по исходному значению, более чем на 5 мс, средний максимальный эффект составил 7,8 мс при приёме дозы 2 мг и 7,2 мс — при дозе 10 мг. Верхняя граница одностороннего 95 % ДИ для ΔΔQTcF во всех временных точках оставалась ниже величины 10 мс. Категориальный анализ не выявил случаев удлинения интервала QTc >480 мс на фоне лечения, случаев удлинения интервала QTc на >60 мс от исходного уровня и случаев увеличения скорректированного или не скорректированного значения QT / QTc >500 мс.
Функция лёгких
Приём одной или нескольких доз препарата в течение 28 дней не сопровождался клинически значимым увеличением сопротивления дыхательных путей при проведении оценки по величине объёма форсированного выдоха за первую секунду (ОФВ1) и мгновенной объёмной скорости выдоха 25–75 % (МОС 25–75) от форсированной жизненной ёмкости лёгких (ФЖЕЛ). При однократном приёме нетерапевтической дозы (>10 мг) отмечена небольшая тенденция к снижению ОФВ1. Приём нескольких доз приводил к лёгким или умеренным изменениям ОФВ1 и МОС 25–75 % вне зависимости от величины дозы и времени суток, которые не сопровождались клиническими проявлениями увеличения сопротивления дыхательных путей.
Одновременный приём сипонимода и пропранолола приводил к минимальному снижению ОФВ1 по сравнению с монотерапией пропранололом. Изменения, отмеченные при применении отдельных препаратов или их комбинаций, оставались в пределах физиологической вариабельности ОФВ1 и не имели клинической значимости.
Фармакокинетика
Всасывание
Время (Tmax) достижения максимальной концентрации сипонимода (Cmax) в плазме крови после многократного приёма внутрь составляло около 4 часов (от 2 до 12 часов). Сипонимод характеризуется значительным всасыванием (≥70 %, исходя из величины радиоактивности в моче и количества метаболитов в фекалиях с экстраполяцией до бесконечности). Абсолютная биодоступность сипонимода при приёме внутрь составляет около 84 %. При приёме сипонимода в дозе 2 мг один раз в сутки в течение 10 дней среднее значение Cmax на десятый день составляло 30,4 нг/мл, а среднее значение AUCtau — 558 ч × нг/мл. Равновесное состояние достигалось приблизительно через 6 дней многократного приёма один раз в сутки.
Приём пищи не влиял на системную экспозицию сипонимода (Сmax и AUC), несмотря на позднее достижение Tmax, которое составляло 8 часов после однократного приёма препарата. В связи с вышесказанным препарат можно принимать вне зависимости от приёма пищи (см. раздел «Способ применения и дозы»).
Распределение
Сипонимод умеренно распределяется в тканях, средний объём распределения составляет 124 л. Доля сипонимода в плазме крови у человека составляет 68 %. В исследованиях у животных показано, что сипонимод легко проходит через гематоэнцефалический барьер. Степень связывания сипонимода с белками плазмы крови у здоровых добровольцев и у пациентов с нарушением функции печени и нарушением функции почек составляет >99,9 %.
Биотрансформация/метаболизм
Сипонимод подвергается активному метаболизму в основном за счёт изофермента CYP2C9 (79,3 %), и в меньшей степени — изофермента CYP3A4 (18,5 %).
Предполагается, что фармакологическая активность основных метаболитов сипонимода, М3 и M17, не вносит вклада в его клиническую эффективность и безопасность у человека. Согласно результатам in vitro исследований, сипонимод при приёме в терапевтической дозе 2 мг 1 раз в сутки и его основные системные метаболиты М3 и М17 не обладают клинически значимым потенциалом межлекарственного взаимодействия со всеми исследованными изоферментами CYP системы цитохрома и белками-переносчиками, в связи с чем проведение соответствующих клинических исследований не требовалось.
Изофермент CYP2C9 является полиморфным, в зависимости от генотипа изменяется доля участия двух путей окислительного метаболизма в выведении препарата. Согласно результатам моделирования физиологически-зависимой фармакокинетики, в зависимости от CYP2C9-генотипа различается степень ингибирования изофермента CYP2C9, и индукции пути, опосредованного изоферментом CYP3A4. Предположено, что чем меньше метаболическая активность изофермента CYP2C9 при соответствующем генотипе, тем более выражен эффект веществ, влияющих на изофермент CYP3A4, на экспозицию сипонимода.
Выведение
У пациентов с PC кажущийся системный клиренс (CL/F) составил 3,11 л/ч. Кажущийся период полувыведения сипонимода из плазмы крови составляет около 30 часов.
Выведение из системной циркуляции происходит в основном за счёт метаболизма и последующего выведения с желчью/калом. В моче сипонимод в неизменённом виде не обнаруживается.
Линейность
Концентрация сипонимода после многократного приёма в дозах от 0,3 мг до 20 мг один раз в сутки увеличивается дозопропорционально.
Концентрация сипонимода в равновесном состоянии в 2–3 раза превышает концентрацию после приёма первой дозы и достигается приблизительно через 6 дней при приёме один раз в сутки. Для достижения клинической терапевтической дозы 2 мг необходим шестидневный период фазы титрации, после чего требуется дополнительно 4 дня приёма для достижения равновесной концентрации в плазме крови.
Фармакокинетика в особых популяциях
Генотип изофермента CYP2C9
Генотип изофермента CYP2C9 влияет на CL/F сипонимода. В двух анализах популяционной фармакокинетики показано, что участники с генотипами CYP2C9*1*1 и *1*2 являются быстрыми метаболизаторами, участники с генотипом *2*2 и *1*3 — промежуточными метаболизаторами, а участники с генотипом *2*3 и *3*3 — медленными. У участников с генотипами CYP2C9*2*2, *1*3, *2*3 и *3*3 отмечено уменьшение показателя CL/F на 20 %, 35–38 %, 45–48 % и 74 % соответственно по сравнению с участниками с генотипом CYP2C9*1*1. Таким образом, экспозиция сипонимода у участников с генотипами CYP2C9*2*2, *1*3, *2*3 and *3*3 увеличивалась приблизительно на 25 %, 61 %, 91 % и 284 % по сравнению с участниками с генотипом *1*1 (см. разделы «Способ применения и дозы» и «Особые указания»).
Пациенты пожилого возраста (65 лет и старше)
Согласно оценке популяционной фармакокинетики, не требуется коррекция дозы сипонимода у пожилых пациентов (в возрасте 65 лет и старше). В клинических исследованиях не участвовали пациенты старше 61 года. Сипонимод следует с осторожностью применять у пожилых пациентов (см. раздел «Способ применения и дозы»).
Пол
Результаты оценки популяционной фармакокинетики свидетельствуют, что коррекция дозы в зависимости от пола не требуется.
Раса / этническая принадлежность
У здоровых добровольцев европеоидной расы и японского происхождения не отмечено различий фармакокинетических параметров после приёма однократной дозы, что говорит об отсутствии этнических различий в фармакокинетике сипонимода.
Нарушение функции почек
Коррекции дозы у пациентов с нарушением функции почек слабой, средней или тяжёлой степени не требуется. Средний период полувыведения и Cmax сипонимода (общего и несвязанного) были сопоставимы у пациентов с нарушением функции почек тяжёлой степени и здоровыми добровольцами. Величина AUC общего и несвязанного вещества были незначительно выше (на 23–33 %) по сравнению со здоровыми добровольцами. Влияние терминальной стадии почечной недостаточности или гемодиализа на фармакокинетику сипонимода не изучалось. Учитывая высокую степень связывания сипонимода с белками плазмы крови (>99,9 %), маловероятно, что гемодиализ может оказывать влияние на концентрацию общего и несвязанного сипонимода, в связи с чем коррекция дозы не представляется необходимой.
Нарушение функции печени
Применение сипонимода у пациентов с нарушением функции печени тяжёлой степени противопоказано. У пациентов с нарушением функции печени лёгкой или средней степени тяжести коррекция дозы не требуется. После приёма однократной дозы 0,25 мг величина AUC несвязанного сипонимода у пациентов с нарушением функции печени средней и тяжёлой степени выше соответственно на 15 % и 50 % по сравнению со здоровыми добровольцами. Средний период полувыведения сипонимода не изменялся при нарушении функции печени.
Показания
Показан для лечения взрослых пациентов с вторично-прогрессирующим рассеянным склерозом (подробную информацию о пациентах см. в разделе «Фармакодинамика»).
Противопоказания
- Повышенная чувствительность к активному веществу или арахису, сое или другим вспомогательным веществам.
- Синдром иммунодефицита.
- Прогрессирующая мультифокальная лейкоэнцефалопатия или криптококковый менингит в анамнезе.
- Активные злокачественные заболевания.
- Нарушение функции печени тяжёлой степени (класс C по классификации Чайлд-Пью).
- Наличие в анамнезе в течение предшествующих 6 месяцев инфаркта миокарда, нестабильной стенокардии, инсульта / транзиторной ишемической атаки, сердечной недостаточности в стадии декомпенсации (требующей стационарной терапии) или сердечной недостаточности класса III/IV по классификации Нью-Йоркской кардиологической ассоциации (см. раздел «Особые указания»).
- Атриовентрикулярная блокада II и III степени типа Мобитц II, синоатриальная блокада, синдром слабости синусового узла в анамнезе при отсутствии электрокардиостимулятора (см. раздел «Особые указания»).
- Гомозиготный генотип изофермента CYP2C9*3 (CYP2C9*3*3) (медленный метаболизатор).
- Беременность и грудное вскармливание, а также у пациенток с сохранённым репродуктивным потенциалом, не использующих контрацепцию.
- Дети до 18 лет (эффективность и безопасность не установлены).
- Непереносимость лактозы, дефицит лактазы, глюкозо-галактозная мальабсорбция.
С осторожностью
- Пациенты старше 65 лет.
- Начало терапии у пациентов с нарушением функции печени слабой и средней степени тяжести.
- Значимое заболевание печени в анамнезе.
- Тяжёлые инфекционные заболевания в стадии обострения до разрешения состояния.
- Одновременное применение с противоопухолевыми, иммуномодулирующими или иммуносупрессивными средствами.
- Начало терапии при одновременном применении бета-адреноблокаторов.
- Сахарный диабет, увеит или сопутствующее заболевание сетчатки в анамнезе.
- Симптоматическая брадикардия или эпизоды синкопе в анамнезе.
- Неконтролируемая артериальная гипертензия.
- Тяжёлый нелеченый синдром апноэ сна.
Применение при беременности и в период грудного вскармливания
Беременность
Женщины с сохранённым репродуктивным потенциалом / контрацепция у женщин
Применение сипонимода противопоказано у женщин с сохранённым репродуктивным потенциалом, не использующих эффективные методы контрацепции. Перед началом терапии препаратом у пациенток с сохранённым репродуктивным потенциалом необходимо получить отрицательный результат теста на беременность, а также проинформировать о серьёзном риске для плода. Пациенткам с сохранённым репродуктивным потенциалом необходимо использовать надёжные методы контрацепции во время лечения сипонимодом, а также как минимум в течение 10 дней после приёма последней дозы препарата.
Необходимо принимать во внимание риск тяжёлого обострения заболевания при прекращении терапии препаратом в связи с планируемой беременностью.
Сведения о применении препарата у беременных женщин крайне ограничены или отсутствуют.
В исследованиях у животных показана эмбриотоксичность и фетотоксичность сипонимода у крыс и кроликов, а также тератогенность у крыс, в том числе увеличение эмбриофетальной смертности и частоты пороков развития скелета и внутренних органов при экспозиции, сравнимой с экспозицией у человека при применении в суточной дозе 2 мг. Кроме того, клинический опыт применения другого модулятора S1P рецепторов во время беременности показал двукратное увеличение риска крупных врождённых пороков развития по сравнению с риском в общей популяции.
Исходя из сказанного, применение сипонимода во время беременности противопоказано. Терапию препаратом необходимо прекратить как минимум за 10 дней до наступления запланированной беременности. Необходимо отменить лечение сипонимодом при наступлении беременности у пациентки, получающей терапию препаратом. Следует предоставить пациентке медицинское заключение о рисках негативного воздействия препарата на плод, а также провести ультразвуковое исследование.
Грудное вскармливание
Резюме рисков
Неизвестно, проникает ли сипонимод или его основные метаболиты в грудное молоко. Пациенткам, получающим препарат Кайендра, следует отказаться от грудного вскармливания.
Фертильность
Нет данных о влиянии сипонимода на фертильность у человека.
Не отмечено влияния на репродуктивные органы самцов у крыс и обезьян или на показатели фертильности у крыс.
Способ применения и дозы
Препарат принимают внутрь, таблетки проглатывают целиком, запивая водой.
Отбор пациентов
Перед началом терапии препаратом необходимо определить генотип экспрессии изофермента CYP2C9 у пациента. Препарат Кайендра противопоказан у пациентов с CYP2C9*3*3-генотипом (см. раздел «Особые указания», а также «Противопоказания»).
Рекомендации для пациентов при переходе с других препаратов, модифицирующих течение PC, на лечение препаратом Кайендра приведены в подразделе «Предшествующая иммуносупрессивная или иммуномодулирующая терапия» раздела «Особые указания».
Фаза титрации
Лечение начинают со стартовой упаковки препарата Кайендра, рассчитанной на 5 дней.
Терапию начинают с дозы 0,25 мг в первый и второй дни терапии с последующим приёмом дозы 0,5 мг однократно в третий день (2 таблетки по 0,25 мг), затем — 0,75 мг однократно в четвёртый день (3 таблетки по 0,25 мг), и 1,25 мг однократно в пятый день (5 таблеток по 0,25 мг) до достижения поддерживающей дозы 2 мг, начиная с шестого дня терапии.
| День терапии | Титрационная доза, мг | Режим титрации | Упаковка |
| День 1 | 0,25 | 1 × 0,25 мг | Стартовая упаковка |
| День 2 | 0,25 | 1 × 0,25 мг | |
| День 3 | 0,50 | 2 × 0,25 мг | |
| День 4 | 0,75 | 3 × 0,25 мг | |
| День 5 | 1,25 | 5 × 0,25 мг | |
| День 6 | 2 | 1 × 2 мг | Упаковка для поддерживающей терапии |
В течение первых шести дней стартовой терапии препарат следует принимать внутрь ежедневно 1 раз в сутки утром, независимо от приёма пищи.
При пропуске приёма очередной дозы в один из шести дней фазы титрации лечение следует начать с новой стартовой упаковки.
Основная целевая популяция
Рекомендованная поддерживающая доза препарата Кайендра составляет 2 мг 1 раз в сутки независимо от приёма пищи.
Рекомендованная поддерживающая доза для пациентов с CYP2C9*1*3- и CYP2C9*2*3-генотипом указана ниже (см. «Особые группы пациентов», фармакогенетика).
Повторное начало терапии при пропуске поддерживающей дозы
В случае пропуска приёма поддерживающей дозы препарата в течение 4 последовательных дней лечение следует начать повторно с новой стартовой упаковки. Приостановление терапии длительностью до трёх дней включительно не требует повторного начала терапии с фазы титрации, лечение в таком случае продолжают в поддерживающей дозе.
Особые группы пациентов
Пожилые пациенты
Применение препарата у пациентов 65 лет и старше не изучено. В клинических исследованиях участвовали пациенты в возрасте до 61 года. В связи с недостаточностью данных по эффективности и безопасности следует с осторожностью применять препарат у пожилых пациентов (см. раздел «Фармакодинамика»).
Фармакогенетика
Рекомендованная поддерживающая доза для пациентов с CYP2C9*1*3- и CYP2C9*2*3-генотипами составляет 1 мг 1 раз в сутки (см. разделы «Особые указания» и «Фармакологические свойства»). У пациентов данной группы лечение начинают с той же стартовой упаковки.
Пациенты с нарушением функции почек
Не требуется коррекции дозы препарата у пациентов данной группы (см. подраздел «Особые группы пациентов» раздела «Фармакологические свойства»).
Пациенты с нарушением функции печени
Препарат противопоказан у пациентов с нарушением функции печени тяжёлой степени (класс C по классификации Чайлд-Пью, см. раздел «Противопоказания»). Несмотря на то, что не требуется коррекции дозы препарата у пациентов с нарушением функции печени лёгкой и средней степени тяжести, следует соблюдать осторожность при начале терапии препаратом у пациентов данной группы (см. подраздел «Особые группы пациентов» раздела «Фармакологические свойства»).
Пациенты младше 18 лет
Препарат противопоказан к применению у пациентов младше 18 лет. Безопасность и эффективность препарата у детей и подростков в возрасте от 0 до 18 лет не установлены. Данные отсутствуют.
Побочное действие
Резюме профиля безопасности
В целом, терапию сипонимодом в суточной дозе минимум 2 мг получили 1737 пациентов с PC. Эти пациенты были включены в два исследования: многоцентровое рандомизированное двойное слепое плацебо-контролируемое исследование III фазы у пациентов с вторичнопрогрессирующим PC и многоцентровое рандомизированное двойное слепое плацебо-контролируемое исследование II фазы с адаптивным режимом дозирования у пациентов с ремиттирующим PC. В исследовании III фазы 1651 пациент был рандомизирован 2 : 1 в группу терапии препаратом в дозе 2 мг 1 раз в сутки или группу плацебо. Медиана продолжительности терапии составила 18 месяцев (от 0 до 37 месяцев). В исследовании II фазы 297 пациентов были рандомизированы в группу терапии исследуемым препаратом в дозах от 0,25 мг до 10 мг или группу плацебо с продолжительностью приёма до 6 месяцев. В исследовании III фазы больший процент пациентов завершили двойную слепую часть исследования с применением исследуемого препарата по сравнению с плацебо (66,7 и 59 % пациентов соответственно).
Наиболее частыми причинами прекращения терапии в группе сипонимода и плацебо были решение пациента или опекуна (10,3 % в группе сипонимода и 13 % в группе плацебо), прогрессирование заболевания (9,1 % в группе сипонимода и 14,8 % в группе плацебо) и нежелательные явления (НЯ, 8,5 % в группе сипонимода и 5,1 % в группе плацебо). Наиболее частыми нежелательными реакциями (HP) в исследовании III фазы были головная боль и артериальная гипертензия.
Нежелательные лекарственные реакции (НЛР), выявленные в клинических исследованиях, определены, прежде всего, на основании исследования III фазы. НЛР перечислены ниже в соответствии с системно-органным классом медицинского словаря для нормативно-правовой деятельности MedDRA. В пределах каждого системно-органного класса НЛР распределены по убыванию частоты возникновения. Для оценки частоты использованы следующие критерии: очень часто (≥1/10), часто (от ≥1/100 до <1/10), нечасто (от ≥1/1000 до <1/100), редко (от ≥1/10000 до <1/1000), очень редко (<1/10000).
| Нежелательные реакции | Сипонимод 2 мг N = 1099, % | Плацебо N = 546, % | Категория частоты |
| Инфекционные и паразитарные заболевания | |||
| Herpes zoster* | 2,5 | 0,7 | Часто |
| Доброкачественные, злокачественные и неопределенные новообразования (включая кисты и полипы) | |||
| Меланоцитарный невус* | 4,9 | 2,9 | Часто |
| Нарушения со стороны системы крови и лимфатической системы | |||
| Лимфопения* | 1,3 | 0,0 | Часто |
| Нарушения со стороны нервной системы | |||
| Головная боль* | 15,2 | 13,9 | Очень часто |
| Головокружение | 6,8 | 4,8 | Часто |
| Судороги* | 1,7 | 0,4 | Часто |
| Тремор* | 1,6 | 0,5 | Часто |
| Нарушения со стороны органа зрения | |||
| Макулярный отек* | 1,8 | 0,2 | Часто |
| Нарушения со стороны сердца | |||
| Брадикардия* | 6,2 | 3,1 | Часто |
| АВ-блокада (I и II степени) | 1,6 | 0,7 | Часто |
| Нарушения со стороны сосудов | |||
| Артериальная гипертензия* | 12,6 | 9,1 | Очень часто |
| Нарушения со стороны желудочно-кишечного тракта | |||
| Тошнота | 6,7 | 3,5 | Часто |
| Диарея | 6,4 | 4,2 | Часто |
| Нарушения со стороны скелетно-мышечной и соединительной ткани | |||
| Боль в конечностях* | 6,3 | 4,0 | Часто |
| Общие расстройства и реакции в месте введения | |||
| Периферический отёк* | 8,1 | 4,4 | Часто |
| Астения | 2,5 | 1,3 | Часто |
| Лабораторные и инструментальные данные | |||
| Увеличение показателей функции печени* | 11,3 | 3,1 | Очень часто |
| Снижение показателей функции лёгких* | 1,5 | 0,5 | Часто |
* — для определения частоты развития были сгруппированы предпочтительные термины.
Описание отдельных нежелательных реакций
Инфекционные заболевания
В клиническом исследовании III фазы у пациентов с вторично-прогрессирующим PC общая частота развития инфекций была сравнима в группе сипонимода и группе плацебо (49,0 % и 49,1 % соответственно). Однако, на фоне терапии сипонимодом было отмечено увеличение частоты развития инфекций, вызванных VZV вирусом (2,5 %) по сравнению с плацебо (0,7 %) (см. раздел «Особые указания»).
В продленной части исследования III фазы в группе сипонимода отмечен один случай развития криптококкового менингита.
Макулярный отёк
У пациентов, получавших сипонимод, макулярный отёк развивался чаще (1,8 %), чем в группе плацебо (0,2 %). И хотя большинство случаев возникло в пределах 3–4 месяцев после начала приёма препарата, часть случаев была зарегистрирована у пациентов, получавших препарат более 6–12 месяцев (см. раздел «Особые указания»), У некоторых пациентов отмечалась нечёткость зрения или снижение остроты зрения, однако у других не было отмечено клинических проявлений, нарушение было диагностировано при плановом офтальмологическом обследовании. В целом, после прекращения приёма препарата отмечалось улучшение или спонтанное разрешение состояния. Оценку риска развития рецидива после возобновления приёма препарата не проводили.
Брадиаритмия
В начале терапии сипонимодом может отмечаться кратковременное снижение ЧСС. а также замедление атриовентрикулярной проводимости (см. раздел «Особые указания»).
У 6,2 % пациентов, получавших сипонимод, отмечено развитие брадикардии по сравнению с 3,1 % пациентов, получавших плацебо; развитие АВ-блокады зарегистрировано у 1,7 % пациентов, получавших лечение препаратом по сравнению с 0,7 % пациентов, получавших плацебо. Максимальное снижение ЧСС отмечено в течение первых 6 часов после приёма.
В начале терапии препаратом отмечается транзиторное дозозависимое снижение ЧСС с последующей стабилизацией на фоне приёма препарата в дозе >5 мг. По сравнению с плацебо частота случаев брадиаритмии (АВ-блокада и синусовая пауза) при применении сипонимода была выше. Большинство случаев АВ-блокады и синусовой паузы отмечены при применении препарата в дозе свыше 2 мг, при этом частота таких явлений была заметно выше при отсутствии фазы титрации. Снижение ЧСС, вызванное сипонимодом, может быть скорректировано применением атропина или изопреналина.
Показатели функции печени
У пациентов с PC на фоне терапии сипонимодом описаны случаи повышения активности печёночных ферментов (в основном повышение активности АЛТ). В исследовании III фазы у пациентов с ВПРС повышение активности печёночных ферментов в группе сипонимода (11,3 %) наблюдалось чаще, чем у пациентов в группе плацебо (3,1 %), в основном за счёт увеличения активности печёночных трансаминаз (АЛТ, ACT и гамма-глутамилтрансферазы (ГГТ)). Большая часть случаев повышения активности печёночных ферментов зарегистрирована в течение 6 месяцев после начала терапии. Возвращение показателей активности АЛТ к норме отмечалось примерно в течение 1 месяца после прекращения приёма сипонимода (см. раздел «Особые указания»).
Артериальное давление
В клиническом исследовании III фазы у пациентов с ВПРС чаще отмечалась артериальная гипертензия в группе сипонимода (12,6 %), чем в группе плацебо (9,0 %). Увеличение систолического и диастолического артериального давления отмечалось на ранней стадии терапии препаратом и достигало максимума приблизительно через 6 месяцев терапии (систолическое — 3 мм рт. ст., диастолическое — 1,2 мм рт. ст.), после чего оставалось стабильным. Указанный эффект сохранялся на фоне продолжающегося лечения.
Судороги
Случаи развития судорог в исследовании III фазы у пациентов с ВПРС отмечались у 1,7 % пациентов, получавших сипонимод, и у 0,4 % пациентов в группе плацебо. Неизвестно, связаны ли эти явления исключительно с течением PC, с терапией сипонимодом или с сочетанием этих двух факторов.
Влияние на дыхательную систему
На фоне терапии сипонимодом отмечалось незначительное снижение объёма форсированного выдоха за 1 секунду (ОФВ1) и диффузионной способности лёгких по монооксиду углерода. В клиническом исследовании III фазы у пациентов с ВПРС на 3 и 6 месяце среднее изменение от исходного значения в группе сипонимода составляло -0,1 л в обеих временных точках, в группе плацебо при этом изменений не было. На фоне хронической терапии данное снижение не приводило к клинически значимым НЯ и не сопровождалось увеличением количества сообщений о развитии кашля или одышки.
Если отмечено ухудшение клинического течения любого из указанных в инструкции побочных эффектов, или Вы заметили любые другие побочные эффекты, не указанные в инструкции, сообщите об этом врачу.
Передозировка
Здоровые добровольцы получали сипонимод как однократно (в дозе от 0,1 до 75 мг), так и многократно (в дозе от 0,25 до 20 мг). На основании случая развития брадикардии с клиническими проявлениями после приёма разовой дозы 75 мг, установлено, что максимальная переносимая доза составляет 25 мг. Максимальная исследованная доза с многократным применением составила 20 мг в течение 28 дней, хорошо переносилась участниками (9 добровольцев получали 100 мг в последний день приёма, 5 — до 200 мг в сутки в течение 3–4 дней). Из 9 участников у некоторых наблюдались преходящие слабо или умеренно выраженные отклонения лабораторных показателей функции печени без клинических проявлений.
После приёма сипонимода в дозе 84 мг у одного из пациентов (с депрессией в анамнезе) не было отмечено каких-либо НЯ, связанных с передозировкой, за исключением небольшого увеличения активности печёночных трансаминаз.
При развитии передозировки при первом приёме препарата или во время фазы титрации следует контролировать состояние пациента с целью выявления вероятных признаков и симптомов брадикардии, с возможностью продления наблюдения до утра следующего дня. Необходимо регулярно контролировать показатели артериального давления, частоты пульса с проведением ЭКГ исследования (см. разделы «Способ применения и дозы» и «Особые указания»).
Специфического антидота для сипонимода не существует. Сипонимод не удаляется в значимом количестве из организма методом диализа или плазмафереза.
Взаимодействие с другими лекарственными средствами
Фармакодинамические взаимодействия
Противоопухолевые препараты, иммуномодуляторы и иммуносупрессанты
Применение препарата Кайендра в комбинации с противоопухолевыми, иммуномодулирующими или иммуносупрессивными препаратами не изучалось. Следует соблюдать осторожность при одновременном применении указанных препаратов с сипонимодом в связи с риском развития кумулятивного влияния на иммунную систему во время терапии, а также в течение нескольких недель после прекращения применения любого из указанных препаратов (см. раздел «Особые указания»).
Замену другой терапии, модифицирующей течение заболевания, на терапию препаратом Кайендра проводят с учётом механизма действия ранее применяемого препарата и периода его полувыведения во избежание развития аддитивного угнетающего эффекта на иммунную систему, вместе с тем стараясь снизить риск реактивации заболевания.
Принимая во внимание механизм действия алемтузумаба, а также описанный в инструкции по его применению иммуносупрессивный эффект, применение препарата Кайендра после курса терапии алемтузумабом не рекомендовано, за исключением случаев, когда ожидаемая польза отчётливо превышает возможный риск у конкретного пациента.
У пациентов, получавших ранее терапию интерфероном-бета или глатирамера ацетатом, лечение препаратом Кайендра может быть начато непосредственно после прекращения применения вышеуказанных препаратов.
Антиаритмические средства, препараты, удлиняющие интервал QT, а также препараты, которые могут снижать ЧСС
В связи с риском кумулятивного влияния на ЧСС не следует начинать терапию препаратом у пациентов, получающих антиаритмические средства IA класса (например, хинидин, прокаинамид) и III класса (например, амиодарон, соталол), препараты, удлиняющие интервал QT, с известными проаритмогенными свойствами, блокаторы кальциевых каналов, снижающие ЧСС (например, верапамил или дилтиазем), или другие препараты, которые могут снижать ЧСС (например, ивабрадин или дигоксин). При рассмотрении вопроса о возможности терапии сипонимодом у пациентов данной категории следует провести консультацию кардиолога (см. подраздел «Рекомендации по началу терапии у пациентов с некоторыми кардиологическими состояниями в анамнезе» раздела «Особые указания»).
Бета-адреноблокаторы
Следует соблюдать осторожность, начиная применение препарата у пациентов, получающих бета-адреноблокаторы, в связи с возможным развитием аддитивного эффекта снижения ЧСС (см. подраздел «Рекомендации по началу терапии у пациентов с некоторыми кардиологическими состояниями в анамнезе» раздела «Особые указания»). Начало терапии препаратом Кайендра возможно у пациентов, получающих стабильные дозы бета-адреноблокатора.
Отрицательное хронотропное действие при одновременном применении сипонимода и пропранолола оценено в ходе специализированного исследования фармакодинамики и безопасности. Добавление пропранолола к терапии сипонимодом на фоне равновесного фармакодинамического/фармакокинетического состояния сопровождалось менее выраженным отрицательным хронотропный действием (чем в случае кумулятивного влияния) по сравнению с добавлением сипонимода к терапии пропранололом в равновесном фармакодинамическом / фармакокинетическом состоянии (кумулятивное влияние на ЧСС).
Вакцинация
Поскольку применение живых аттенуированных вакцин может увеличивать риск развития инфекций, следует избегать иммунизации живыми аттенуированными вакцинами на фоне терапии препаратом и в течение 4 недель после её завершения (см. подраздел «Вакцинация» раздела «Особые указания»).
Во время терапии препаратом, а также в течение 2 месяцев после прекращения лечения сипонимодом вакцинация может быть менее эффективной. Считается, что эффективность вакцинации не снижается при прекращении терапии сипонимодом за 1 неделю до планируемой вакцинации и возобновлении приёма препарата не ранее 4 недель после её проведения (см. подраздел «Вакцинация» раздела «Особые указания»).
Фармакокинетические взаимодействия
Вероятность влияния других препаратов на фармакокинетику сипонимода (сипонимод в качестве субстрата)
Метаболизм сипонимода осуществляется в основном за счёт изофермента CYP2C9 (79,3 %), и в меньшей степени — за счёт изофермента CYP3A4 (18,5 %) системы цитохрома P450. Изофермент CYP2C9 является полиморфным ферментом, и в зависимости от CYP2C9-генотипа можно спрогнозировать результат лекарственного взаимодействия в присутствии веществ, метаболизируемых изоферментом CYP3A ИЛИ CYP2C9.
Ингибиторы изоферментов CYP2C9 и CYP3A4
Не рекомендовано одновременное применение сипонимода с препаратами, вызывающими умеренное ингибирование изофермента CYP2C9, и умеренное или мощное двойное ингибирование изоферментов CYP2C9/CYP3A4 в связи со значительным увеличением экспозиции сипонимода. В состав такой одновременной терапии могут входить умеренный двойной ингибитор изофермента CYP2C9/CYP3A4 (например, флуконазол) или умеренный ингибитор изофермента CYP2C9 с одновременным применением умеренного или мощного ингибитора изофермента CYP3A4.
Применение флуконазола (умеренного ингибитора изоферментов CYP2C9/CYPJA4) В дозе 200 мг в сутки в равновесном состоянии с сипонимодом однократно в дозе 4 мг у здоровых добровольцев с CYP2C9*1*1-генотипом приводило к двукратному увеличению AUC последнего препарата.
Согласно результатам оценки потенциала лекарственного взаимодействия с использованием моделирования физиологически-зависимой фармакокинетики, при одновременном применении сипонимода с любым типом ингибитора изофермента CYP3A4 или CYP2C9 у пациентов всех генотипов ожидается максимально двукратное увеличение AUC сипонимода, за исключением генотипа CYP2C9*2*2. В присутствии умеренного ингибитора изофермента CYP2C9/CYP3A4 у пациентов с генотипом CYP2C9*2*2 ожидается увеличение AUC сипонимода в 2,7 раз.
Индукторы изоферментов CYP2C9 и CYP3A4
Возможно одновременное применение препарата Кайендра с большинством индукторов изоферментов CYP3A4 и CYP2C9.
Тем не менее, учитывая ожидаемое снижение экспозиции сипонимода, следует соблюдать осторожность при одновременном применении:
- с мощными индукторами изофермента CYP3A4 / умеренными индукторами изофермента CYP2C9 (например, карбамазепином) у всех пациентов вне зависимости от генотипа;
- с умеренными индукторами изофермента CYP3A4 (например, модафинилом) у пациентов с CYP2C9*1*3- или -*2*3-генотипами (см. раздел «Фармакологические свойства»).
Согласно результатам оценки потенциала лекарственного взаимодействия с использованием моделирования физиологически-зависимой фармакокинетики, ожидается значительное уменьшение экспозиции сипонимода (на 76 % и 51 % соответственно). Одновременное применение сипонимода в суточной дозе 2 мг на фоне ежедневного приёма рифампицина в дозе 600 мг (мощный индуктор изофермента CYP3A4 и умеренный индуктор изофермента CYP2C9) у участников исследования с генотипом CY2C9*1*1 отмечалось уменьшение AUCtau,ss и Сmax сипонимода соответственно на 57 % и 45 %.
Вероятность влияния сипонимода на фармакокинетику или фармакодинамику других препаратов
Оральные контрацептивы
При одновременном применении не выявлено клинически значимого влияния сипонимода на фармакокинетику или фармакодинамику комбинированных оральных контрацептивов, содержащих этинилэстрадиол и левоноргестрел. Таким образом, на фоне терапии сипонимодом эффективность исследуемого орального контрацептива сохранялась.
Несмотря на отсутствие исследований лекарственного взаимодействия с иными оральными контрацептивами, содержащими другие прогестагены, не ожидается влияния сипонимода на их экспозицию.
Лабораторные исследования
Поскольку сипонимод уменьшает количество лимфоцитов в крови за счёт их перераспределения в периферические лимфоидные органы, для оценки различных популяций лимфоцитов у пациентов, получающих лечение препаратом Кайендра, не может использоваться подсчёт количества лимфоцитов в периферической крови. При проведении тестов с циркулирующими мононуклеарами у пациентов, получающих сипонимод, требуется получение большего объёма крови вследствие снижения количества циркулирующих лимфоцитов.
Особые указания
Инфекционные заболевания
Ключевым фармакодинамическим эффектом препарата является дозозависимое уменьшение количества лимфоцитов в периферической крови до 20–30 % от исходного количества, обусловленное их обратимым перераспределением в лимфоидных тканях (см. раздел «Фармакологические свойства»).
В связи с влиянием препарата на иммунную систему возможно увеличение риска развития инфекций (см. раздел «Фармакологические свойства»).
Перед началом терапии препаратом следует получить результат общеклинического анализа крови с лейкоцитарной формулой, выполненного в течение 6 месяцев, предшествующих началу терапии, или после отмены предшествующей терапии. Периодически во время терапии рекомендовано выполнять развёрнутый анализ крови. При подтверждённом снижении абсолютного количества лимфоцитов <0,2 × 109/л дозу препарата следует уменьшить, поскольку в клинических исследованиях дозу препарата снижали у пациентов с абсолютным количеством лимфоцитов <0,2 × 109/л. При подтверждённом снижении абсолютного количества лимфоцитов до <0,2 × 109/ л у пациента, получающего препарат в дозе 1 мг, терапию препаратом следует временно прервать. При восстановлении абсолютного количества лимфоцитов ≥0,6 × 109/л возможно рассмотреть возобновление лечения препаратом. У пациентов с тяжёлым инфекционным заболеванием в активной фазе необходимо отложить начало лечения препаратом до разрешения данного состояния.
Поскольку такой остаточный фармакодинамический эффект, как уменьшение количества лимфоцитов в периферической крови, может сохраняться в течение 3–4 недель после прекращения терапии, в этот период необходимо сохранять насторожённость в отношении развития инфекций (см. подраздел «Прекращение терапии препаратом»).
Пациентов, получающих терапию препаратом, следует проинформировать о необходимости незамедлительно сообщать врачу о симптомах инфекции. При развитии симптомов инфекционного процесса на фоне терапии препаратом необходимо провести эффективные диагностические и терапевтические мероприятия. При развитии серьёзной инфекции у пациента, получающего препарат Кайендра, следует рассмотреть возможность отмены терапии.
В продлённой фазе клинического исследования препарата зарегистрирован случай развития криптококкового менингита. Случаи развития криптококкового менингита зарегистрированы при применении другого модулятора S1P-рецепторов. Врачу следует сохранять насторожённость в отношении возможных симптомов криптококкового менингита. При развитии симптомов, позволяющих заподозрить развитие данного состояния, следует незамедлительно провести соответствующие диагностические мероприятия. Применение препарата Кайендра следует временно приостановить до исключения криптококкового менингита, при подтверждении диагноза следует начать соответствующее лечение.
В программе разработки препарата не отмечено случаев развития прогрессирующей мультифокальной лейкоэнцефалопатии (ПМЛ), однако такие случаи зарегистрированы при применении другого модулятора S1Р-рецепторов. Врачу следует сохранять насторожённость в отношении возможных клинических симптомов или данных МРТ, позволяющих заподозрить ПМЛ. При подозрении на ПМЛ следует приостановить лечение препаратом Кайендра до исключения данного диагноза.
В программе разработки препарата зарегистрированы случаи герпес-вирусной инфекции, (включая один случай реактивации вируса Varicella zoster (VZV) с развитием менингита, вызванного VZV). Пациентов, у которых отсутствуют документально подтвержденные данные о перенесённой ветряной оспе или о полном курсе вакцинации против VZV, следует обследовать для выявления антител к VZV перед началом терапии.
Вакцинация
При отсутствии антител к вирусу VZV пациенту следует провести полный курс вакцинации до начала терапии препаратом, которое следует отложить на 1 месяц до развития полного иммунного ответа на вакцинацию.
Следует избегать применения живых аттенуированных вакцин во время терапии препаратом, а также в течение 4 недель после её прекращения.
Во время терапии препаратом вакцинация может быть менее эффективной. Рекомендовано прервать терапию сипонимодом за 1 неделю до планируемой вакцинации, возобновлять применение препарата следует не ранее, чем через 4 недели после неё.
Противоопухолевые препараты, иммуномодуляторы и иммуносупрессанты
Учитывая риск аддитивного влияния на иммунную систему, следует соблюдать осторожность при одновременном применении препарата с противоопухолевыми средствами, иммуномодуляторами или иммунодепрессантами (в том числе глюкокортикостероидами) (см. раздел «Взаимодействие с другими лекарственными средствами»).
Макулярный отек
В клиническом исследовании III фазы случаи развития макулярного отёка отмечались чаще при применении сипонимода (1,8 %) по сравнению с плацебо (0,2 %). Большинство этих случаев зарегистрированы в первые 3–4 месяца после начала терапии. Поскольку такие случаи отмечались и при более длительном применении препарата, пациента следует проинформировать о необходимости сообщать лечащему врачу о нарушениях со стороны зрения в течение всего времени приёма препарата. В этих случаях рекомендовано провести осмотр глазного дна, включая макулярную область. Терапию препаратом не следует начинать у пациентов с макулярным отёком до разрешения данного состояния. У пациентов с сахарным диабетом, увеитом в анамнезе, а также у пациентов с сопутствующими поражениями сетчатки отмечается повышенный риск развития макулярного отёка. У пациентов данных категорий рекомендуется проводить офтальмологическое обследование до начала и во время терапии препаратом Кайендра.
Оценку возможности продления терапии препаратом у пациентов с макулярным отеком не проводили. Вопрос о возможности прекращения терапии препаратом следует рассматривать в индивидуальном порядке на основании отношения польза-риск.
Брадиаритмия
ЧСС
В связи с развитием транзиторного снижения ЧСС на фоне начала терапии сипонимодом, лечение препаратом начинают по схеме поэтапного повышения дозы до достижения поддерживающей дозы к шестому дню терапии (см. раздел «Способ применения и дозы»).
Снижение ЧСС начинается в титрационной фазе в течение часа после приёма первой дозы и достигает максимума в первый день в течение приблизительно 3–4 часов. В последующие дни титрационной фазы на фоне продолжающегося повышения дозы отмечается дальнейшее уменьшение ЧСС с максимальным снижением к пятому-шестому дню от исходного уровня, наблюдавшегося в первый день. Максимальное суточное снижение ЧСС в абсолютном среднечасовом значении наблюдается в первый день, при этом частота пульса снижается в среднем на 5–6 ударов в минуту (уд/мин). В последующие дни терапии пост-дозовые снижения менее выражены. На фоне последующего приёма ЧСС начинает увеличиваться после шестого дня и достигает уровня плацебо в течение десяти дней от начала терапии.
Случаи снижения ЧСС менее 40 уд/мин отмечались редко. В целом, пациенты с брадикардией не испытывали симптомов, небольшое количество пациентов отмечали слабо или умеренно выраженные симптомы, включая головокружение или слабость, которые разрешались самостоятельно без вмешательства в течение 24 часов. При необходимости для устранения снижения ЧСС, индуцированного сипонимодом, возможно парентеральное введение атропина или изопреналина.
Атриовентрикулярная проводимость
Начало терапии препаратом сопровождалось транзиторный замедлением атриовентрикулярной проводимости, временные рамки были аналогичны снижению ЧСС в фазе титрации. В большинстве случаев отмечены явления АВ-блокады I степени (удлинение интервала PR на электрокардиограмме, ЭКГ). В клинических исследованиях у 1,7 % пациентов в начале терапии отмечено развитие АВ-блокады II степени, как правило, по типу Мобитц I. Явления нарушения проводимости, как правило, были транзиторными, асимптомными, разрешались в течение 24 часов и не требовали отмены препарата.
Рекомендации по началу терапии у пациентов с некоторыми кардиологическими состояниями в анамнезе
В качестве меры предосторожности у пациентов с синусовой брадикардией (ЧСС <55 уд/мин), АВ-блокадой первой или второй степени (Мобитц I), или инфарктом миокарда в анамнезе, или сердечной недостаточностью (пациенты с сердечной недостаточностью I или II степени по классификации Нью-Йоркской кардиологической ассоциации) в анамнезе следует проводить наблюдение в течение 6 часов после приёма первой дозы препарата с целью выявления признаков и симптомов брадикардии. Рекомендовано проведение ЭКГ исследования перед началом терапии и по окончании периода наблюдения. При развитии постдозовой брадиаритмии или симптомов, связанных с нарушением проводимости, или при выявлении на ЭКГ, снятой через 6 часов после приёма первой дозы, новых признаков АВ-блокады второй степени или выше, или увеличения интервала QTc ≥500 мс, следует начать соответствующее лечение и продлить наблюдение вплоть до разрешения состояния / восстановления показателей. При необходимости лекарственной терапии необходимо продлить наблюдение по меньшей мере до утра следующего дня и повторить 6-часовое наблюдение после приёма второй дозы. В связи с риском развития серьёзных нарушений сердечного ритма или значимой брадикардии препарат не следует применять у пациентов с симптоматической брадикардией или эпизодами синкопе в анамнезе, неконтролируемой артериальной гипертензией или тяжёлым нелеченым синдромом апноэ сна. У таких пациентов возможность применения сипонимода следует рассматривать только в случае, если ожидаемая польза превышает потенциальные риски. Перед началом терапии у таких пациентов следует проконсультироваться с кардиологом с целью выбора наиболее оптимального способа мониторинга сердечной деятельности.
В специализированном QT-исследовании не выявлено значимого прямого влияния препарата Кайендра на удлинение интервала QT, а также аритмогенного потенциала, связанного с удлинением интервала QT. Начало терапии препаратом может приводить к уменьшению ЧСС и косвенно — к удлинению интервала QT в фазе титрации. Применение препарата не изучалось у пациентов со значимым удлинением интервала QТ (>500 мс), а также у пациентов, получающих препараты, способные удлинять интервал QT. При рассмотрении вопроса о терапии препаратом у пациентов с сопутствующим значимым удлинением интервала QT или у пациентов, получающих препараты, способные удлинять интервал QT, с общеизвестным аритмогенным потенциалом, перед началом терапии следует проконсультироваться с кардиологом с целью выбора наиболее оптимального способа мониторинга сердечной деятельности при старте терапии.
Применение препарата у пациентов с аритмией, требующей медикаментозной коррекции антиаритмическими препаратами класса IA (например, хинидин, прокаинамид) или класса III (например, амиодарон, соталол) не изучалось. Применение антиаритмических препаратов класса IA и класса III у пациентов с брадиаритмией ассоциировано со случаями развития нарушений сердечного ритма типа torsades de pointes. Поскольку начало терапии препаратом Кайендра приводит к снижению ЧСС, не рекомендовано одновременное применение вышеуказанных препаратов в стартовой фазе терапии.
Опыт применения препарата ограничен у пациентов, получающих одновременную терапию препаратами блокаторами кальциевых каналов, снижающими ЧСС (например, верапамил или дилтиазем), или другими средствами, которые могут снижать ЧСС (например, ивабрадин или дигоксин). Одновременное применение данных препаратов во время терапии препаратом Кайендра может привести к развитию тяжёлой брадикардии и блокады сердца. В связи с возможным аддитивным эффектом на ЧСС применение препарата у пациентов, получающих данные средства, в целом не рекомендовано. У таких пациентов возможность применения сипонимода следует рассматривать только в случае, если ожидаемая польза превышает потенциальные риски. При рассмотрении вопроса о возможности одновременного применения указанных препаратов на фоне стартовой терапии препаратом Кайендра следует проконсультироваться с кардиологом с целью определения возможности перехода на терапию препаратами, не снижающими ЧСС, или выбора оптимального мониторинга сердечной деятельности в стартовой фазе терапии.
Брадиаритмический эффект более выражен при применении препарата одновременно с бета-адреноблокаторами. У пациентов, получающих бета-адреноблокаторы в стабильных дозах, следует предварительно оценить ЧСС в состоянии покоя. Применение препарата Кайендра на фоне хронического приёма бета-адреноблокаторов возможно, если ЧСС в покое >50 уд/мин. В случае, если ЧСС в покое ≤50 уд/мин, следует прервать приём бета-адреноблокатора до восстановления ЧСС >50 уд/мин, после чего следует начать применение препарата Кайендра. Возобновление терапии бета-адреноблокатором в таком случае возможно после окончания фазы титрации и достижения поддерживающей дозы препарата Кайендра.
Пропуск приёма и возобновление терапии после перерыва
В случае пропуска приёма одной дозы препарата на протяжении первых шести дней титрационной фазы или при пропуске четырёх и более последовательных суточных доз препарата в поддерживающей фазе, лечение препаратом следует начать по схеме фазы титрации. В этом случае следует придерживаться тех же рекомендаций и схем мониторинга состояния пациента (см. выше подраздел «Рекомендации по началу терапии»).
Функция печени
Перед началом терапии препаратом следует получить результаты лабораторного определения показателей активности трансаминаз и концентрации билирубина (т.е. выполненных в течение шести месяцев, предшествующих началу терапии). В клиническом исследовании III фазы у 5,6 % пациентов, получавших сипонимод в дозе 2 мг, и 1,5 % пациентов, получавших плацебо, отмечалось увеличение активности аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (ACT) в три раза превышающих верхнюю границу нормы (ВГН). В клинических исследованиях применение препарата прекращали при увеличении показателей >3 ВГН и наличии симптомов, связанных с функцией печени.
При появлении во время применения препарата симптомов, позволяющих заподозрить нарушение функции печени, таких как необъяснимая тошнота, рвота, боль в животе, повышенная утомляемость, анорексия, сыпь с эозинофилией или желтуха и/или тёмное окрашивание мочи, следует определить активность ферментов печени и, при выявлении серьёзного повреждения печени, препарат следует отменить.
Несмотря на отсутствие данных о повышенном риске увеличения активности печёночных ферментов у пациентов с существующими заболеваниями печени на фоне терапии препаратом Кайендра, следует соблюдать осторожность при применении препарата у пациентов со значимыми заболеваниями печени в анамнезе.
Новообразования кожи
В клиническом исследовании наиболее частым кожным новообразованием являлась базальноклеточная карцинома кожи, которая была с одинаковой частотой зарегистрирована как в группе пациентов, получавших терапию препаратом (1,01 %, 12 пациентов), так и получавших плацебо (1,23 %, 7 пациентов). Однако у пациентов, получавших лечение сипонимодом, а также длительную терапию другим модулятором S1Р-рецепторов, отмечалось развитие других злокачественных новообразований кожи, включая меланому. Пациентов, получающих лечение препаратом, следует предупредить о необходимости применения средств защиты от солнечного излучения. У таких пациентов не следует проводить одновременную фототерапию с использованием УФ-В излучения или ПУВА-терапию.
Непредвиденные неврологические или психиатрические симптомы/признаки
При применении другого модулятора S1P-рецепторов отмечены редкие случаи развития синдрома обратимой задней энцефалопатии, которые не зарегистрированы в программе разработки препарата Кайендра. Однако, в случае развития любых непредвиденных неврологических или психиатрических симптомов или признаков (например, когнитивное расстройство, изменение поведения, кортикальные зрительные нарушения или любые другие кортикальные неврологические симптомы / явления или любые другие, позволяющие заподозрить повышения внутричерепного давления) или при стремительном ухудшении неврологического статуса на фоне терапии препаратом следует немедленно провести полную оценку физического и неврологического статуса и рассмотреть возможность проведения МРТ.
Предыдущая терапия иммуносупрессорами и иммуномодуляторами
Замену другой терапии, модифицирующей течение заболевания, на терапию препаратом Кайендра проводят с учётом механизма действия ранее применяемого препарата и периода его полувыведения во избежание развития аддитивного угнетающего эффекта на иммунную систему, вместе с тем стараясь снизить риск реактивации заболевания. Перед началом терапии сипонимодом рекомендовано получить результаты развёрнутого анализа крови с подсчётом лимфоцитов, выполненного после отмены предшествующей терапии с целью убедиться в разрешении эффектов предыдущей терапии относительно иммунной системы (например, цитопении).
Учитывая механизм действия и иммунодепрессивный эффект алемтузумаба, описанный в инструкции по его применению, применение препарата Кайендра после прекращения терапии алемтузумабом не рекомендовано, за исключением случаев, когда ожидаемая польза явно превышает возможный риск для конкретного пациента. В целом начало терапии сипонимодом возможно непосредственно после окончания предшествующей терапии β-интерфероном или глатирамера ацетатом
Влияние на артериальное давление
Из клинических исследований исключили пациентов с артериальной гипертензией, не поддающейся медикаментозному контролю. Для пациентов с неконтролируемой артериальной гипертензией необходима специализированная медицинская помощь.
В клиническом исследовании III фазы артериальная гипертензия чаще отмечалась у пациентов с ВПРС, получавших сипонимод (12,6 %), чем получавших плацебо (9,0 %).
Применение сипонимода приводило к повышению систолического и диастолического артериального давления вскоре после начала лечения с максимальным эффектом после шестого месяца терапии (систолическое на 3 мм рт. ст., диастолическое — на 1,2 мм рт. ст.) и последующей стабилизацией. Данный эффект сохранялся на фоне продолжающейся терапии. Во время лечения препаратом необходимо регулярно контролировать артериальное давление.
Фармакогенетика
Перед началом терапии препаратом необходимо провести генотипирование по изоферменту CYP2C9 с целью определения CYP2C9-метаболического статуса у пациента. Препарат Кайендра противопоказан у пациентов, гомозиготных по аллелю CYP2C9*3 (распространенность CYP2C9*3*3-генотипа составляет приблизительно 0,3–0,4 % у европеоидов и менее среди других рас). Приём препарата у пациентов данной категории приводит к значительному увеличению концентрации сипонимода в плазме крови (см. раздел «Фармакологические свойства»).
Во избежание увеличения экспозиции сипонимода у пациентов с CYP2C9*2*3- или CYP2C9*1*3-генотипами рекомендованная поддерживающая доза препарата составляет 1 мг в сутки (см. раздел «Способ применения и дозы» и «Фармакологические свойства»).
Прекращение терапии препаратом
После окончания терапии сипонимод остаётся в кровотоке до десяти дней. Применение других препаратов в этот период приведёт к увеличению экспозиции сипонимода.
У большинства пациентов с ВПРС (90 %) количество лимфоцитов возвращается к исходному в течение 10 дней после прекращения терапии сипонимодом. Однако, в течение 3–4 недель после приёма последней дозы препарата могут наблюдаться остаточные фармакодинамические эффекты, такие как уменьшение количества периферических лимфоцитов. Применение иммуносупрессантов в этот период может приводить к развитию дополнительного угнетающего воздействия на иммунную систему, в связи с чем следует соблюдать осторожность в течение 3–4 недель после приёма последней дозы сипонимода.
Вспомогательные вещества
Лекарственная форма содержит соевый лецитин. Препарат не следует применять у пациентов с гиперчувствительностью к арахису или сое (см. раздел «Противопоказания»). Препарат также не следует применять у пациентов с редкими наследственными нарушениями, связанными с непереносимостью галактозы, тяжёлой лактазной недостаточностью или глюкозо-галактозной мальабсорбцией.
Влияние на способность управлять транспортными средствами, механизмами
Сипонимод оказывает минимальное влияние на способность к управлению транспортными средствами или не оказывает его вовсе. Однако, на фоне начала терапии препаратом может развиваться головокружение. В связи с вышесказанным в первый день терапии пациентам следует отказаться от управления транспортными средствам или механизмами.
Форма выпуска
Таблетки, покрытые плёночной оболочкой, 0.25 мг, 2 мг.
Таблетки, покрытые плёночной оболочкой, 0,25 мг:
по 12 таблеток, покрытых плёночной оболочкой, в блистер из ПА/Ал/ПВХ и алюминиевой фольги. По 1 блистеру в картонном пенале вместе с инструкцией по медицинскому применению в картонную пачку («стартовая упаковка»);
или
по 10 блистеров вместе с инструкцией по медицинскому применению в картонную пачку.
Таблетки, покрытые плёночной оболочкой, 2 мг:
по 14 таблеток, покрытых плёночной оболочкой, в блистер из ПА/Ал/ПВХ и алюминиевой фольги. По 2 блистера вместе с инструкцией по медицинскому применению в картонную пачку.
Допускается наличие контроля первичного вскрытия на картонной пачке.
Хранение
При температуре от 2 до 8 °C.
Препарат следует хранить в недоступном и невидном для детей месте.
Срок годности
2 года.
Препарат не применять по истечении срока годности.
Условия отпуска из аптек
Отпускают по рецепту
Производитель
Novartis Farmaceutica, S.A., Испания
Novartis Pharma Stein, AG, Швейцария
Siegfried Barbera, S.L., Испания
Наименование и адрес юридического лица, на имя которого выдано регистрационное удостоверение
«Новартис Оверсиз Инвестментс АГ», Лихтштрассе 35, 4056 Базель, Швейцария / Novartis Overseas Investments AG, Lichtstrasse 35, 4056 Basel, Switzerland
Производитель
Производство готовой лекарственной формы
«Новартис Фарма Штейн АГ» / Novartis Pharma Stein AG, Schaffhauserstrasse, 4332 Stein, Switzerland
Первичная, вторичная упаковка и выпускающий контроль качества
«Новартис Фармасьютика С.А.», / Novartis Farmaceutica S.A., Ronda de Santa Maria, 158, Barbera del Valles, 08210 Barcelona, Spain
Получить дополнительную информацию о препарате, а также направить
Претензии и информацию о нежелательных явлениях можно по следующему адресу в России:
ООО «Новартис Фарма»
125315, г. Москва, Ленинградский проспект, дом 72, корпус 3
тел. (495) 967 12 70;
факс (495) 967 12 68.